原位乳液聚合制备P(St-co-BMA)/GO复合材料
2014-03-26邢妍
邢 妍
(上海交通大学化学化工学院,上海市 200240)
石墨烯是一种新型的平面片层材料,石墨烯片层由sp2杂化的碳原子构成,具有高模量、高强度、高导电性和较大的比表面积[1-2]。将石墨烯添加到聚合物中能够提高聚合物的力学性能、导电性能和导热性能等,但石墨烯的平面大π键结构使单片层的石墨烯易在聚合物中发生堆叠而团聚在一起,而原位聚合可有效解决该问题[3]。Wang Xin等[4]通过原位聚合法制备了聚氨酯/石墨烯复合材料,当石墨烯质量分数为2%时,复合材料的拉伸断裂强度和储能模量分别提高239%,202%,具有良好的导电性和热稳定性。氧化石墨烯(GO)表面具有多种含氧官能团,可参与聚合,用于制备原位接枝的聚合物,以提高石墨烯在聚合物中的分散性。Xu Zhen等[5]利用GO片层上的含氧官能团与己内酰胺缩聚合,聚合过程中将GO还原,制备了石墨烯接枝聚酰胺6复合材料。GO在水及众多有机溶剂中分散性良好,常被用于原位聚合制备聚合物的前驱体,基于GO的原位乳液聚合常被用于制备聚合物胶乳。Morimune等[6]利用乳液聚合法制备了聚甲基丙烯酸甲酯(PMMA)/GO复合材料,当w(GO)为1%时,复合材料的杨氏模量由纯PMMA的2.2 MPa增至4.1 MPa。Patole等[7]利用原位乳液聚合法制备了具有良好导电性的聚苯乙烯(PS)/石墨烯复合材料,并研究了复合材料的微观形态结构。
本工作采用原位乳液聚合法制备苯乙烯(St)-甲基丙烯酸丁酯(BMA)共聚物[P(St-co-BMA)]/GO复合材料。为了改善PS的脆性,加入第二单体BMA,使P(St-co-BMA)具有良好的韧性和可塑性,而GO可有效改善其力学性能。
1 实验部分
1.1 原料
天然石墨,粒径44 µm,纯度为99.8%,阿法埃莎(中国)公司生产。BMA,分析纯,TCI(上海)公司生产。St;浓硫酸,纯度98%;高锰酸钾;过氧化氢水溶液,纯度30%;盐酸,纯度70%;十二烷基磺酸钠(SDS);引发剂,过硫酸铵;Al2(SO4)3;无水乙醇:均为国药集团化学试剂有限公司生产。
1.2 GO的制备
采用改良的Hummers方法[8]。称取定量天然石墨,缓慢加入浓硫酸,在冰浴中搅拌均匀;向烧杯中缓慢加入高锰酸钾,在室温(23 ℃)条件下搅拌24 h;此后,向体系中缓慢加入去离子水,直至没有气泡产生,加入过氧化氢水溶液;过滤,洗涤数次后冻干,即得到GO。
1.3 P(St-co-BMA)/GO复合材料的制备
称取定量GO,分散在水中,充分搅拌后经超声振动,制备质量浓度为1 mg/mL的GO溶液。称取定量SDS于三口烧瓶中,在水或GO溶液(GO含量为单体总质量的1%)中溶解,搅拌30 min至SDS溶解,向烧瓶中通氮气15 min;待气流稳定后,按m(St)/m(BMA)分别为70∶30,50∶50,30∶70向体系加入St,BMA混合单体;将过硫酸铵溶液滴加到烧瓶中;在油浴中加热烧瓶,至温度达到85 ℃,在200 r/min的搅拌速率下反应6 h;反应完全后,向乳液中先后加入10 mL质量分数为5%的Al2(SO4)3溶液和10 mL稀盐酸破乳,过滤和洗涤固体产物,烘干备用。复合材料在200 ℃条件下模压成型。
1.4 分析与测试
将制备的P(St-co-BMA)乳液及P(St-co-BMA)/GO 复合材料稀释后滴在硅片上,在空气中干燥,喷金后在美国FEI 公司生产的Nano450型场发射扫描电子显微镜上观察乳胶粒的微观结构。将稀释的乳液滴在微栅上,干燥后在日本Jeol公司生产的 JEM-2100F型透射电子显微镜下观察其形貌。热重(TG)分析在美国PE公司生产的TGA205型热重分析仪上进行,氮气气氛,升温速率为20℃/min,温度40~800 ℃。差示扫描量热法(DSC)分析在美国PE公司生产的Pyris1型差示扫描量热分析仪上进行,氮气气氛,升温速率为20 ℃/min,温度-80~200 ℃。力学性能采用美国Instron公司生产的Instron 4465型拉力机按ASTM D 412—2006测试,拉伸速度为10 mm/min。
2 分析与讨论
2.1 P(St-co-BMA)及其复合材料的形貌
由图1a可知:P(St-co-BMA)乳胶粒子呈圆球状,颗粒大小分布均匀,粒径为40~100 nm。由图1b可知:P(St-co-BMA)/GO 复合材料中几乎没有单独存在的P(St-co-BMA)乳胶粒子,P(Stco-BMA)乳胶粒子均吸附在GO片层表面。GO具有平面片层结构,其平面大π键结构与P(St-co-BMA)中St单元产生π-π堆叠作用,使P(St-co-BMA)乳胶粒子易于吸附在GO的表面[9]。通过透射电子显微镜(TEM)观察,进一步验证了P(Stco-BMA)的微观形态。由图1c可知:P(St-co-BMA)乳胶粒子呈圆球状,而P(St-co-BMA)/GO复合材料中P(St-co-BMA)乳胶粒子吸附在GO片层表面(见图1d),这结果与扫描电子显微镜(SEM)测试结果一致。
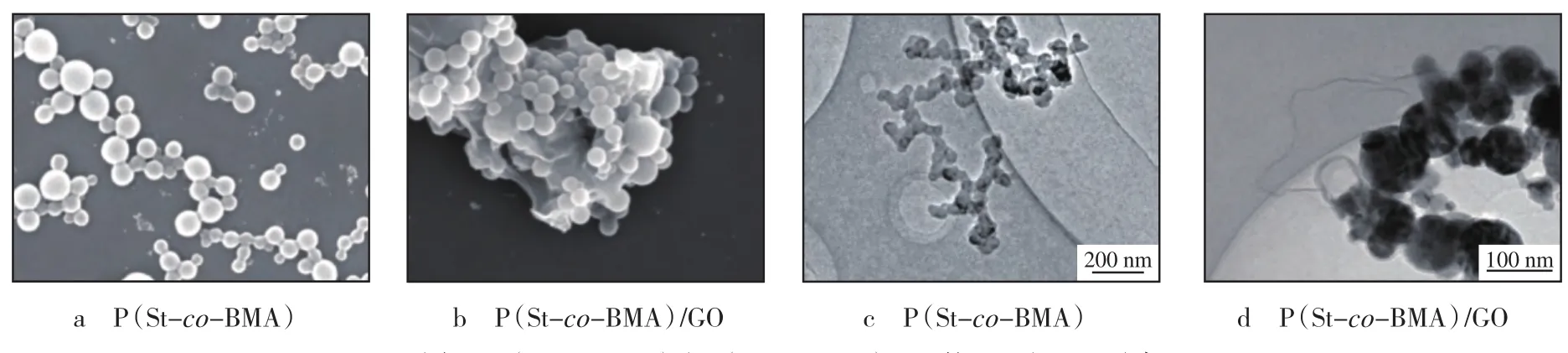
图1 P(St-co-BMA)和P(St-co-BMA)/GO 的SEM和TEM照片Fig.1 SEM and TEM photos of P(St-co-BMA) and P(St-co-BMA)/GO
2.2 P(St-co-BMA)及其复合材料的热性能
由图2可知:随着BMA单体含量的不断增加,P(St-co-BMA)的玻璃化转变温度(tg)明显降低。P(St-co-BMA)中St链段为脆性结构单元,BMA为柔性结构单元,随着BMA结构单元含量的增加,tg相应降低。添加GO后,与相同m(St)/m(BMA)条件下制备的P(St-co-BMA)相比,P(St-co-BMA)/GO复合材料的tg更低。由此推测,GO增大了BMA的竞聚率,使P(St-co-BMA)中BMA单元的含量增加,因此,P(St-co-BMA)/GO 复合材料的tg降低。
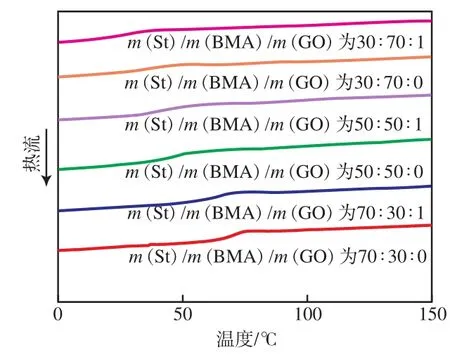
图2 P(St-co-BMA)和P(St-co-BMA)/GO 的DSC曲线Fig.2 DSC curves of P(St-co-BMA) and P(St-co-BMA)/GO
以m(St)/m(BMA)为70∶30为例,由图3可知:P(St-co-BMA)的起始热分解温度为297 ℃(质量损失5%时的温度),450 ℃时分解完全;P(St-co-BMA)的微分热重(DTG)曲线有两个峰,分别在360,410 ℃处;加入GO后,P(St-co-BMA)/GO 复合材料的起始热分解温度升到326℃,DTG曲线只有一个峰,最大分解速率对应的温度为412 ℃。其他m(St)/m(BMA)条件下制备的P(St-co-BMA)及其复合材料具有相似的特征,即P(St-co-BMA)/GO复合材料的起始热分解温度远高于P(St-co-BMA),且其DTG曲线均只有一个峰出现。
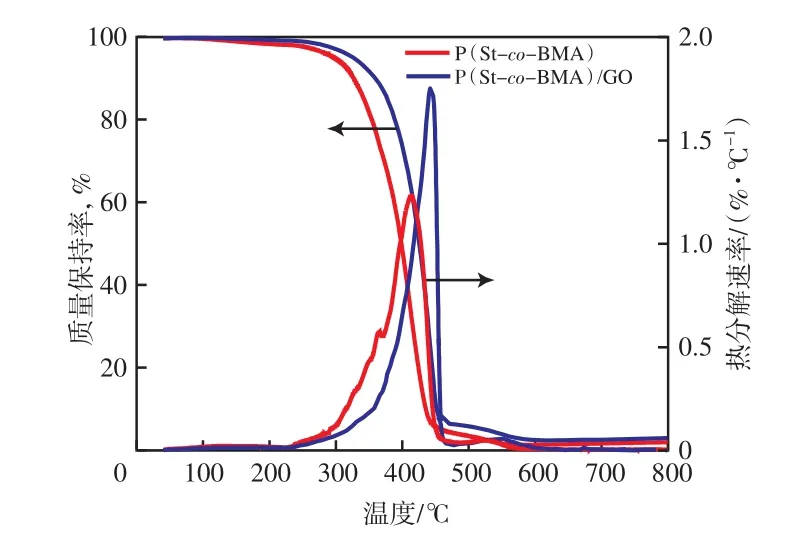
图3 P(St-co-BMA)和P(St-co-BMA)/GO 的TG和DTG曲线Fig.3 TG and DTG curves of P(St-co-BMA) and P(St-co-BMA)/GO
2.3 P(St-co-BMA)及其复合材料的力学性能
由表1可知:随着BMA含量的增加,P(St-co-BMA)的拉伸强度明显下降。当m(St)/m(BMA)为30∶70时,P(St-co-BMA)的拉伸强度只有9.5 MPa,但断裂伸长率明显提高,达239%。添加GO后,与相同m(St)/m(BMA)条件下制备的P(Stco-BMA)相比,P(St-co-BMA)/GO复合材料的拉伸强度都有所提高。当m(St)/m(BMA)为30∶70时,P(St-co-BMA)/GO复合材料的拉伸强度升至23.7 MPa,比P(St-co-BMA)提高近250%。与P(St-co-BMA)相比,P(St-co-BMA)/GO 复合材料的断裂伸长率并没有明显降低。综上所述,GO有效地提高了P(St-co-BMA)的拉伸强度,同时对其断裂伸长率的影响不大。
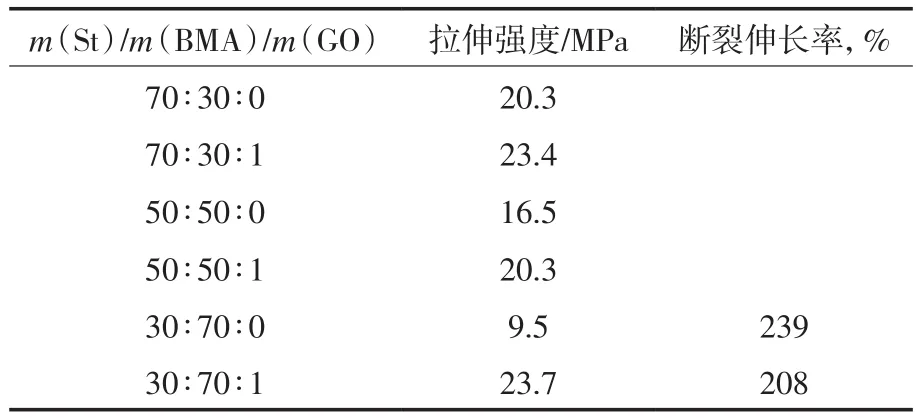
表1 P(St-co-BMA)和P(St-co-BMA)/GO 的力学性能Tab.1 The mechanical properties of P(St-co-BMA) and P(St-co-BMA)/GO
3 结论
a)随着BMA单体含量的增加,P(St-co-BMA)的tg明显降低。添加GO后,与相同m(St)/m(BMA)条件下制备的P(St-co-BMA)相比,P(St-co-BMA)/GO复合材料的tg更低。
b)P(St-co-BMA)/GO复合材料的起始热分解温度远高于P(St-co-BMA),且其DTG曲线均只有一个峰出现。
c)随着BMA含量的不断增加,P(St-co-BMA)的拉伸强度明显下降。添加GO后,与相同m(St)/m(BMA)条件下制备的P(St-co-BMA)相比,P(St-co-BMA)/GO复合材料的力学性能都有所提提高。当m(St)/m(BMA)为30∶70时,P(Stco-BMA)/GO复合材料的拉伸强度由P(St-co-BMA)的9.5 MPa升至23.7 MPa,提高近250%,且断裂伸长率没有明显降低。
[1] Bolotin K I, Sikes K J, Jiang Z, et al. Ultrahigh electron mobility in suspended graphene[J]. Solid State Communications, 2008,146(9/10): 351-355.
[2] Balandin A A, Ghosh S, Bao Wenzhong, et al. Superior thermal conductivity of single-layer graphene[J]. Nano Letters, 2008, 8(3): 902-907.
[3] Kim H, Miura Y, Macosko C W. Graphene/polyurethane nanocomposites for improved gas barrier and electrical conductivity[J]. Chemistry of Materials, 2010, 22(11): 3441-3450.
[4] Wang Xin, Hu Yuan, Song Lei, et al. In situ polymerization of graphene nanosheets and polyurethane with enhanced mechanical and thermal properties[J]. Journal of Materials Chemistry, 2011, 21(12): 4222-4227.
[5] Xu Zhen,Gao Chao. In situ polymerization approach to graphene-reinforced nylon-6 composites[J]. Macromolecules,2010, 43(16): 6716-6723.
[6] Morimune S, Nishino T, Goto T. Ecological approach to graphene oxide reinforced poly(methyl methacrylate) nanocomposites[J].ACS Applied Materials & Interfaces, 2012, 4(7): 3596-3601.
[7] Patole A S, Patole S P, Kang H, et al. A facile approach to the fabrication of graphene/polystyrene nanocomposite by in situ microemulsion polymerization[J]. Journal of Colloid and Interface Science, 2010, 350(2): 530-537.
[8] Bai Xin, Wan Chaoying, Zhang Yong, et al. Reinforcement of hydrogenated carboxylated nitrile-butadiene rubber with exfoliated graphene oxide[J]. Carbon, 2011, 49(5): 1608-1613.
[9] Che Man S H, Thickett S C, Whittaker M R, et al. Synthesis of polystyrene nanoparticles "armoured" with nanodimensional graphene oxide sheets by miniemulsion polymerization[J].Journal of Polymer Science Part A: Polymer Chemistry, 2013,51(1): 47-58.
