常染色体显性遗传性多囊肾家系PKD1、PKD2基因突变分析
2014-03-21骆杰伟孟晓嵘郑星宇魏世超张雪梅范丁前
骆杰伟 孟晓嵘 郑星宇 魏世超 胡 丹 杨 笑 张雪梅 范丁前
常染色体显性遗传性多囊肾病(ADPKD)是很常见的单基因遗传性疾病,是终末期肾病(ESRD)的最常见的遗传病因[1],在全世界范围内有0.12亿人发病[2];中国肾移植科学登记系统数据显示,囊性肾脏病(3.5%)占中国ESRD第四位病因[3]。目前至少发现3个基因与本病有关,已明确的是定位于16p13.3的PKD1基因和定位于4q21的PKD2基因,分别编码多囊蛋白1(PC1)和多囊蛋白2(PC2) 。与PKD1突变有关的患者至少占85%,而与PKD2有关的≤15%;尚有可能不明确的第三种基因与之有关。PKD1患者较PKD2患者病情重,发病早[4,5]。此研究试图用华大基因(BGI)外显子序列捕获技术平台检测一个多囊肾家系突变方式。
对象与方法
研究对象本家系先证者来自于福建省立医院住院患者,44岁男性,汉族,临床表现为腰酸、肉眼血尿、腹胀、高血压、泌尿系感染、双肾多发结石、胸腔积液等。并收集本家系相关成员共8个血样及临床资料,其中包括先行收集的已故的先证者母亲Ⅱ4,而先证者妹妹(40岁)Ⅲ5在同期表现出上述症状。多囊肾诊断参考Ravine等[6]提出超声诊断:(1)<30岁患者单侧或双侧有≥2个囊肿;(2)30~59岁患者双侧肾囊肿≥2个;(3)≥60岁患者双侧肾囊肿≥4个;(4)如果具有家族史或多囊肝等肾外表现或基因检测,诊断标准可适当放宽。
经医院医学伦理委员会同意,受试者均签署知情同意书。
研究方法
临床表型检测根据先证者及家系成员临床表现行肝肾彩超、电子计算机X射线断层扫描技术(CT)或核磁共振等检测,采集血、尿常规、生化全套等。
基因组DNA的提取抽取先证者15 ml外周血及家系其他7名成员抽取2 ml外周血,按试剂盒(Omega E.Z.N.A.® Tissue DNA Kit)说明书方法提取基因组DNA。
Illumina测序[7,8]及生物信息学分析[9](1)样品检测:先证者DNA样品由Nanodrop 2000检测浓度,取3 μg以上DNA样品用于DNA片段化。(2)样品打断:用Covaris法将DNA片段打断至250 bp左右的片段并回收。(3)Illumina测序文库构建:将回收片段进行末端修复反应,将测序特定接头(Adapter)同末端修复产物进行连接反应,根据Adapter上的通用引物结合位点进行产物的扩增反应,纯化回收产物。(4)目的基因的捕获和富集:用集合了多种基因的DNA捕获芯片进行包括目的基因PKD1、PKD2在内的多个基因片段进行富集,富集产物用Illumina 2000(Hiseq2000测序仪)进行测序反应。(5)数据分析:原始图像文件经过Illumina base calling Software 1.7进行碱基读取,获得读长为90 bp双末端序列(reads)。去除低质量和污染的reads,去除接头Adapter序列得到纯化数据进行序列比对分析,用soap软件分析拷贝数、多态性(SNP)和插入/缺失(INDEL),并且进行注释筛选可疑致病突变。并经SIFT(http://sift.jcvi.org/)和Polyphen软件(http://genetics.bwh.harvard.edu/pph/)对其进行蛋白功能预测。上述部分由深圳华大基因研究院协作按照其操作标准完成。
Sanger测序验证 (1)引物合成:PKD1基因序列来自GenBank(NM_000296),PKD1基因10号外显子引物测序来自文献[10]。引物由北京六合华大基因科技股份有限公司合成。上游序列:5′-G ̄C ̄A ̄G ̄G ̄C ̄A ̄G ̄T ̄T ̄G ̄G ̄G ̄C ̄A ̄T ̄C ̄T ̄C ̄T ̄G-3′,下游序列:5′-G ̄A ̄C ̄C ̄C ̄T ̄G ̄G ̄G ̄C ̄A ̄G ̄C ̄A ̄G ̄A ̄C ̄A ̄G-3′。(2)PCR产物扩增:25 μl的反应体系中包含10×扩增缓冲液2.5 μl,dNTP (2.5 mmol/l) 2 μl,Forward primer(3 mmol/L)3 μl,Reverse primer(3 mmol/L)3 μl,DNA模板1 μl,Ex Taq 0.2 μl,H2O 18.3 μl。在PCR仪(PTC-200 PCR,BIO-RAD)上94℃变性5 min,按照94℃变性40s,57℃退火40s,72℃延伸60s条件循环35次后72℃延伸10 min。(3)PCR产物的纯化和测序:PCR产物采用Omega公司E.Z.N.A.TMGel Extraction Kit按照操作说明纯化,测序按照BigDye Terminator v1.1试剂盒PCR产物标准流程操作。(4)DNA测序结果分析:采用DNAMAN Version 5.2.2与正常序列比对。当怀疑杂合缺失或插入时,将PCR产物连接在PGEM-T Easy(Promega)载体上挑选亚克隆进行测序。
结 果
临床资料经临床调查,发现此家系有4个发病者(图1);先证者(Ⅲ3)以“腰酸胀、排肉眼血尿、尿频、尿痛、尿频”为主因就诊,既往发现“多囊肾、多囊肝”10年,入院前5年曾于外院行“双侧多囊肾去顶术”;已故母亲死于“肝肾联合移植术”,术前诊断“多囊肾、多囊肝、肝功能不全、尿毒症”,先证者妹妹(Ⅲ5)也发现“多囊肾、多囊肝”10年,3年前行“左侧多囊肾去顶术”,肾功能正常;经查先证者血肌酐104 μmol/L,尿素痰6.9 mmol/L,血尿酸392 μmol/L,谷丙转氨酶15 U/L,谷草转氨酶22 U/L,谷氨酰转移酶54 U/L,碱性磷酸酶46.0 U/L,总胆红素12.94 μmol/L,胱抑素1.42 mg/L,糖类抗原199(Ca199)93.73 U/ml(0.0~27.0 U/ml),D-二聚体1.48 μg/ml,血红蛋白139 g/L,电解质、甲状腺功能正常;尿常规:白细胞++++,蛋白++++,潜血++++;心电图:窦性心动过缓;彩超:双肾多发结石,多囊肝,多囊肾,前列腺增生伴结石,胆囊、胰腺及腹膜显示不清;血压波动于110~130/80~95 mmHg;磁共振(MRI)示:肝脏及双肾轮廓明显增大,其内可见弥漫多发大小不一类圆形囊性灶,最大径约9.2 cm,以长T1、长T2信号为主,双肾内部分病灶呈T1信号。肝、肾实质明显受压显示不清,以肝左外叶残余可见的肝实质较多。邻近组织明显受压移位或扩
张受限。扫描野内双侧胸腔少量积液,呈长T2信号。考虑为多囊肝、多囊肾(部分肾囊肿合并出血),双侧胸腔少量积液。此家系发病者影像均表现出肾外症状如多囊肝比多囊肾症状重(图2)。
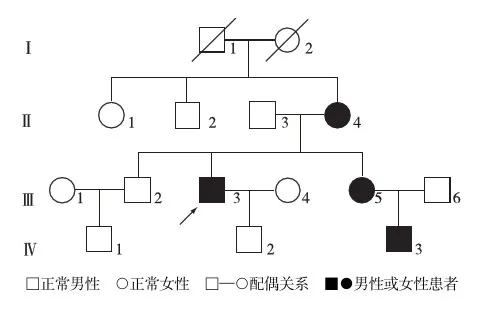
图1 常染色体显性遗传性多囊肾家系图
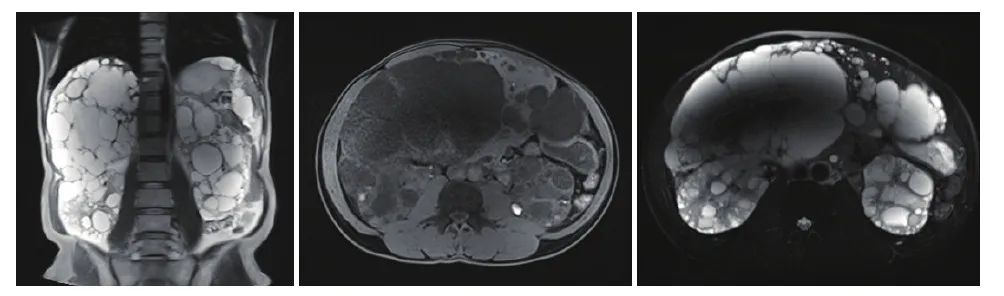
图2 先证者多囊肾、多囊肝磁共振影像图
PKD1、PKD2基因外显子捕获测序质量先证者的PKD1、PKD2基因外显子捕获测序质量报告图表可见,除PKD1基因第1号外显子外,其余外显子的测序覆盖均>99%(图3)。各外显子的平均测序深度,与测序深度中位数均比较接近,说明测序的随机性比较好。
PKD1、PKD2基因外显子突变分析对先证者的PKD1、PKD2基因外显子测序深度数据进行统计学分析后,未发现存在大片缺失或重复。在PKD1基因编码区发现1个框移突变c.2085_2086insC(p.Ala696Argfs17X),为杂合子;该突变造成PKD1基因氨基酸编码提前终止,很可能影响其蛋白功能;其在人群中发生频率极低。在PKD1基因编码区还发现两个错义突变杂合子:4340C>T(p.Ala1447Val)、2216A>G (P.Arg739Gln),经SIFT和Polyphen对其进行蛋白功能预测,结果为无害,为已知多态性位点;及同义变异:1119T>C(p.Leu373Leu)、2670C>T (p.Asn890Asn),其中p.Leu373Leu为为已知的多态性位点。在PKD2基因上未发现框移、无义、剪切、错义或同义变异位点。
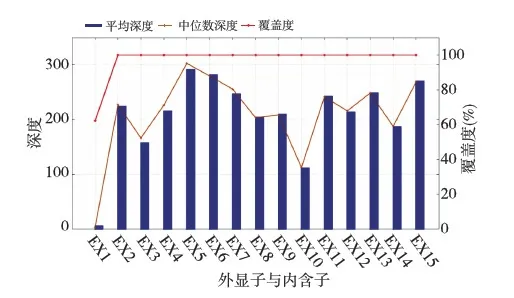
图3 PKD1基因外显子捕获测序质量报告图表
家系成员经Sanger测序,Ⅱ4、Ⅲ3、Ⅲ5、Ⅳ3均查出p.Ala696Argfs17X突变方式,而在Ⅱ3、Ⅲ2、Ⅳ1、Ⅳ2未查出此突变方式。因此,推测PKD1基因编码区的框移突变c.2085_2086insC(p.Ala696Argfs17X)为此家系的致病性突变。
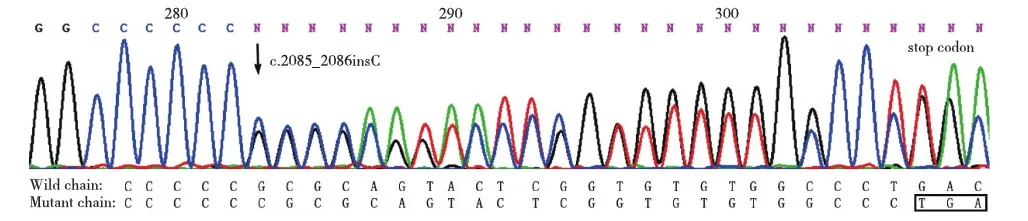
图4 常染色体显性遗传性多囊肾先证者PKD1基因exon 10测序图
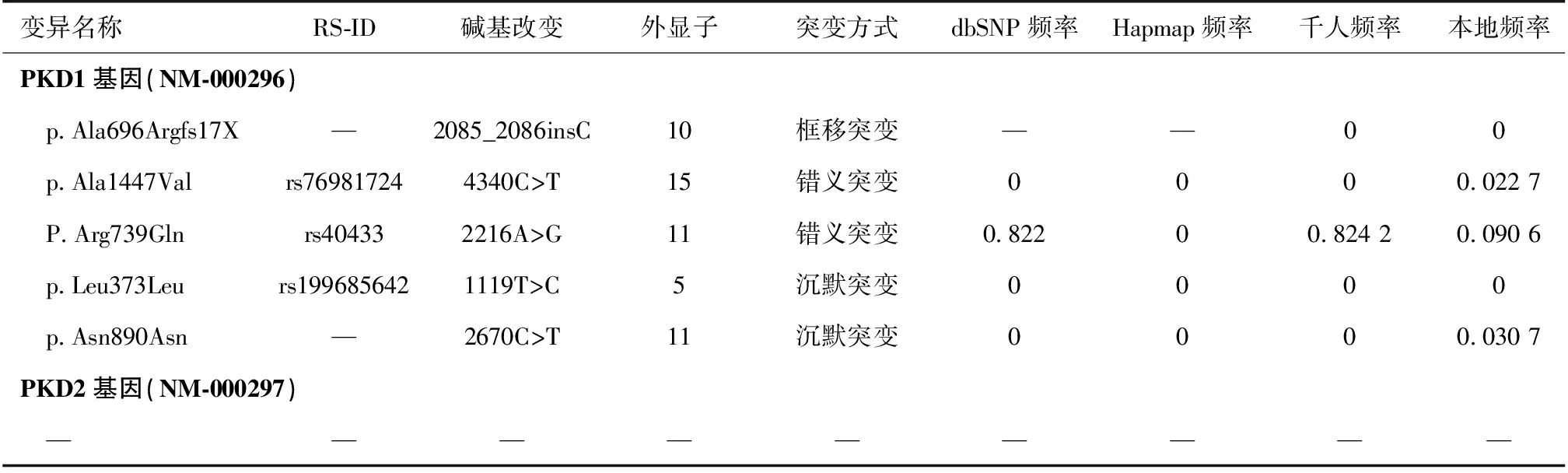
表1 常染色体显性遗传性多囊肾先证者的突变方式
dbSNP频率:dbSNP数据库中收录的关于此SNP的频率信息;Hapmap频率:Hapmap数据库中收录的关于此SNP在亚裔人种中的频率信息;千人频率:千人基因组计划中全部测序样本中关于此SNP的频率信息;本地频率:华人基因(BGI)收集的>625个正常人测序样本中关于SNP的频率信息
讨 论
人PKDl基因长度约52 kb,含有46个外显子,编码的PC1是4 302个氨基酸残基构成的11次跨膜的糖蛋白,由细胞外区(NH2端)、跨膜区与细胞内区(COOH端)组成,细胞内区含有螺旋-螺旋交互结构,PC1分布细胞膜初级纤毛及与细胞连接处如黏附斑、桥粒等位置[11]。PC1参与细胞-细胞或细胞-基质交互作用,PC1通过海胆卵胶受体(REJ)、G蛋白耦联、Wnt等信号传导途径挥发作用[5],PKD2基因长度为68 kb,含有15个外显子,编码的PC2,表达于内质网、细胞膜底外侧和初级绒毛中,含6个跨膜区域,其NH2端和COOH端均位于细胞内,为非选择性阳离子通道,能运载钙离子;PC2的C端可能存在一个特定的盘绕功能域,与PC1相连形成多囊蛋白复合物[12]。PC1与PC2配合,进一步激活钙离子通道,共同启动由细胞外向细胞内信号传导,调控其他下游活性基因。ADPKD发病涉及多条信号传导通路,这些通路异常可引起囊泡上皮细胞异常增殖与囊液过度分泌而导致囊泡发展。PKD1和PKD2基因如发现突变,或可导致PC1与PC2结构与功能发生改变,信号传导通路异常,引起细胞异常增殖和囊液过度分泌而引起多囊肾与多囊肝。
搜索ADPKD突变数据库(Version 3.0,http://pkdb.mayo.edu/),PKD1基因有2 324个突变点,明确的致病突变有868个,包括错义突变263个、框移突变449个、插入或缺失43个、大片缺失或重复20个及剪切突变93个; PKD2基因有278个突变点,其中致病突变有162个,包括错义突变48个、框移突变75个、插入或缺失2个、大片缺失或重复6个及剪切突变31个。目前发现PKD1基因的大多突变分布在35~46外显子单拷贝区,而5′近端区域1~34外显子因存在同源基因多次核苷酸重复拷贝,给DNA测序及突变检测带来难度[13]。应用第二代测序技术平台提高了检出率,PKD1、PKD2编码区、多拷贝区和单拷贝区检出真阳性率可分别达69.4%、50% 和100%[14];此多囊肾家系应用二代测序外显子序列捕获技术,检测出5′近端区域区多个突变,其中p.Ala696Argfs17X参与了此多囊肾家系的发病,其余突变位点经检索均为多态性位点。该突变造成PKD1基因氨基酸编码提前终止,其必然翻译出被截短、有缺陷的蛋白质,很可能影响其蛋白功能;此突变导致的ADPKD至今已发现有10个左右家系[15,16],分布在不同人种。另外,这个家系病情较重,一个原因可能是p.Ala696Argfs17X位于5′近端区域,Rossetti等[17]曾报道了80个家系的324例ADPKD患者,发现致病突变点在5′端的患者临床症状较3′端重,而进入ESRD的年龄与发生的突变位点类型无关联,5′近端和3′端区域突变的患者进入ESRD的平均年龄分别为53和56岁(P=0.025),到60岁肾功能尚良好的比例分别为18.9%和39.7%。
特定基因的大片段区域的传统测序主要通过步行PCR法后进行毛细管测序的。但其目标片段>500 kb时,PCR难度及测序成本较高。而单基因病的研究采用的是家系连锁定位,最后定位区域也需要以兆(M)bp来计算的,进一步研究难度也大。近年,发展起来的第二代测序技术,基于边合成边测序(Sequencing by Synthesis)原理,结合基因芯片和新一代测序方法的二项高通量测序技术,产生了新的经济实惠测序技术,如目标序列捕获、全外显子组捕获技术。现有的二代高通量测序平台有:llumina Solexa HiSeq2000、Genome AnalyzerIIx、ABI SOLID4、Roche 45;其中Illumina/Solexa Genome Analyzer应用的是合成法测序,测序用不同颜色的荧光标记4种不同的dNTP,合成互补链时,每添加一种dNTP就会释放出不同的荧光,再用计算机软件分析这些捕捉的荧光信号,获得DNA的序列信息。全外显子组捕获测序可以作为传统家系定位方法的有益补充,将极大的推动遗传疾病的研究。人类大概85%的单基因疾病突变位点位于外显子区域,新一代测序与传统全基因组测序相比,覆盖度更广、数据准确性更高,更加简便、经济、高效。可应用于寻找复杂疾病致病基因和易感基因等的研究[18]。
目前关于多囊肾发病学说主要有:二次打击学说、螺旋区-螺旋区相互作用、终止信号假说、齐-杂合子学说及纤毛致病学说等。我们推测这些基因突变或引起PC1功能改变,进而可能引起初级纤毛功能障碍,导致无法将感应正常尿流率产生的终止信号传递给肾小管细胞,造成肾小管细胞增殖失控,囊肿不断扩大[19]。其他具有基因多态性位点特征的错义或突变可能在多囊肾的后天的发生发展过程中起着协同作用,值得关注。
(感谢深圳华大基因临床检验中心给予的技术支持)
1Drenth JP,Martina JA,van de Kerkhof R,et al.Polycystic liver disease is a disorder of cotranslational protein processing.Trends Mol Med,2005,11(1):37-42.
2Helal I.Autosomal dominant polycystic kidney disease:new insights into treatment.Saudi J Kidney Dis Transpl,2013,24(2):230-234.
3Liu ZH.Nephrology in china.Nat Rev Nephrol,2013,9(9):523-528.
4Hateboer N,v Dijk MA,Bogdanova N,et al.Comparison of phenotypes of polycystic kidney disease types 1 and 2.European PKD1-PKD2 Study Group.Lancet,1999,353(9147):103-107.
5Bastos AP,Onuchic LF.Molecular and cellular pathogenesis of autosomal dominant polycystic kidney disease.Braz J Med Biol Res,2011,44(7):606-617.
6Ravine D,Gibson RN,Walker RG,et al.Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1.Lancet,1994,343(8901):824-827.
7He J,Wu J,Jiao Y,et al.IgH gene rearrangements as plasma biomarkers in Non- Hodgkin’s lymphoma patients.Oncotarget,2011,2(3):178-185.
8Wu J,Matthaei H,Maitra A,et al.Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development.Sci Transl Med,2011,3(92):92ra66.
9Li R,Li Y,Kristiansen K,et al.SOAP:short oligonucleotide alignment program.Bioinformatics,2008,24(5):713-714.
10 Tan YC,Michaeel A,Blumenfeld J,et al.A novel long-range PCR sequencing method for genetic analysis of the entire PKD1 gene.J Mol Diagn,2012,14(4):305-313.
11 Ibraghimov-Beskrovnaya O,Bukanov N.Polycystic kidney diseases:from molecular discoveries to targeted therapeutic strategies.Cell Mol Life Sci,65(4):605-619.
12 Tsiokas L,Kim E,Arnould T,et al.Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2.Proc Natl Acad Sci USA,1997,94(13):6965-6970.
13 Watnick TJ,Gandolph MA,Weber H,et al.Gene conversion is a likely cause of mutation in PKD1.Hum Mol Genet,1998,7(8):1239-1243.
14 Qi XP,Du ZF,Ma JM,et al.Genetic diagnosis of autosomal dominant polycystic kidney disease by targeted capture and next-generation sequencing:utility and limitations.Gene,2013,516(1):93-100.
15 Rossetti S,Hopp K,Sikkink RA,et al.Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing.J Am Soc Nephrol,2012,23(5):915-933.
16 Audrézet MP,Cornec-Le Gall E,Chen JM,et al.Autosomal dominant polycystic kidney disease:comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients.Hum Mutat,2012,33(8):1239-1250.
17 Rossetti S,Burton S,Strmecki L,et al.The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease.J Am Soc Nephrol,2002,13(5):1230-1237.
18 黄建锋,肖华胜.目标序列捕获技术及其应用.生物产业技术,2011,(4):64-69.
19 Kotsis F,Boehlke C,Kuehn EW.The ciliary flow sensor and polycystic kidney disease.Nephrol Dial Transplant,2013,28(3):518-526.
