巨噬细胞与肾脏疾病
2014-03-21任加发综述戴春笋审校
任加发 周 阳 综述 戴春笋 审校
巨噬系统属于单核吞噬细胞系统,其在维持组织内微环境稳态、组织结构重塑及机体免疫调节方面有着非常重要的作用。巨噬细胞(Mφ)在肾脏疾病中的作用一直受到广泛地重视。研究表明Mφ在肾脏疾病中的作用较为复杂,一方面肾组织中Mφ可能是促进损伤和肾脏纤维化的重要致病因素,另一方面在免疫或非免疫介导的肾脏疾病中,Mφ也积极参与了损伤组织的清除并且促进组织结构的恢复。本文就Mφ的生物学特性及在肾脏疾病中的作用等方面进行重点阐述。
Mφ的起源、分型及功能
循环单核细胞起源于骨髓干细胞。在特定细胞因子[如粒细胞-巨噬细胞集落刺激因子(GM-CSF)、巨噬细胞集落刺激因子(M-CSF)]刺激下,骨髓干细胞分化为前单核细胞并进入血液分化为成熟的单核细胞。在损伤组织中,Mφ主要有两个来源:循环中的炎性单核细胞和组织内的固有Mφ。上述由两种不同途径进入组织的Mφ,即M0型Mφ。一般认为炎症组织中聚集的单核巨噬细胞(MO/Mφ)多是从血液中招募而来,但也有研究显示固有Mφ也可通过自身增殖来增加数量[1](图1)。
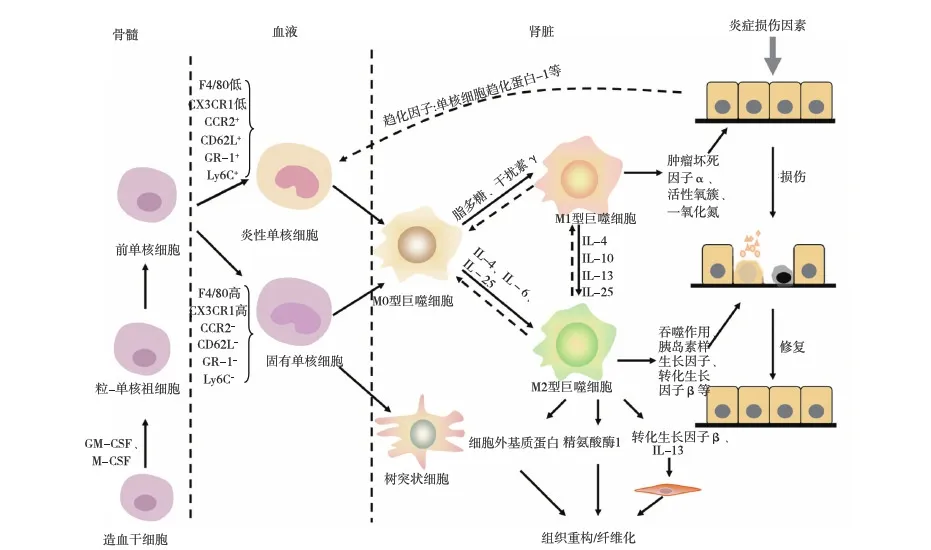
图1 巨噬细胞起源、分化及肾脏损伤、修复、纤维化中的作用示意图
M0型Mφ在不同因子刺激下,分化为不同的亚型。如干扰素γ(IFN-γ)/Toll样受体(TLR)配体促进Mφ分化为经典M1型;而M2型Mφ是包含了除经典M1型以外的各种活化形式的Mφ。M1型高表达致炎因子,促进Th1型免疫反应。相反,M2型主要与抑制寄生虫,促进组织修复、纤维化及免疫调节有关。M2型Mφ又被进一步细分为M2a、M2b和M2c三个亚群。M2型组成及功能复杂并且具有潜在的治疗作用,这几年逐渐成为研究的热点。与M2a型相比,M2c型更加有效地改善肾小球硬化、肾小管萎缩及间质纤维化等[2]。并且不同来源的Mφ,如骨髓和脾脏来源的Mφ对受损的肾脏作用也不尽相同。越来越多的研究着眼于Mφ表面受体及胞内信号通路对Mφ分化的影响,进而研究Mφ与组织损伤、修复及纤维化等之间的关系[3,4]。除了自身分泌的活性因子直接作用于器官实质细胞外,Mφ也通过其他免疫细胞发挥作用。
另外,分化为M1或M2型的Mφ不会单纯地表现为一种状态,在特定条件下它们可以直接相互转化,且有可能回到静息状态,继而被诱导至另一种活化状态[5]。
上述分型是人为地将Mφ从功能上进行简化区分,便于理解。然而,和体外培养相比,Mφ在肾组织内不仅受到炎性介质和细胞因子等影响,细胞与细胞之间也存在着相互作用,因此与上述分型相匹配的Mφ亚群是否真正存在于肾组织中尚存在争议[6],Anders和Ryu[7]认为在体内,可根据疾病损伤修复愈合的不同阶段将Mφ按功能分为促炎型、抗炎型、促纤维化型及纤维溶解型四种类型。
单核巨噬细胞的促炎、分化及增殖机制
单核巨噬细胞的促炎机制Mo/Mφ通过激活免疫系统产生促炎反应,其中涉及丝裂原活化蛋白激酶(MAPK)、核因子κB(NF-κB)、Fcγ受体(FcγR)等多种促炎信号通路。(1)MAPK信号通路:在多种肾小球肾炎模型中,抑制c-Jun N端激酶(JNK) MAPK信号通路虽不能抑制Mφ浸润,但可降低蛋白尿和系膜增生,并且减少iNOS和肿瘤坏死因子α(TNF-α) mRNA的表达[8]。Mφ可诱导足细胞nephrin和podocin表达下降,并且此过程依赖于Mφ的JNK信号通路。在人类或实验性肾小球肾炎模型中,浸润Mφ中的p38 MAPK信号通路被激活。抑制p38 MAPK可减轻Mφ所致肾脏损伤[9]。(2)NF-κB信号通路:浸润Mφ中存在NF-κB的活化,尤见于新月体中。在人类肾小球肾炎中,肾小球NF-κB的激活与促炎细胞因子的产生和组织学损伤有关。抑制NF-κB活化可减轻抗肾小球基膜(GBM)肾炎的蛋白尿和肾脏损伤,减少Mφ及炎性因子分泌(IL-1β、TNF-α)。用显性负相NF-κB抑制蛋白α亚基(I κBα)转化骨髓源性Mφ,可致其产生抗炎表型。尽管发生转化的细胞仅占浸润Mφ总数的10%~15%,但输入这种转化细胞后,急性抗GBM肾炎大鼠的尿蛋白及肾损伤均明显减轻,提示即便是少量抗炎Mφ也能显著抑制内源性Mφ的促炎效应[10],该结论在小鼠阿霉素肾病模型中也得到证实[11]。(3)Fcγ受体信号通路:Mφ表达IgG和IgA的Fc段受体,包括两型FcγR,即活化型和抑制型。缺失活化型FcγRγ链的小鼠,其抗GBM肾炎肾脏损伤减轻[12],而缺失抑制型FcγRIIB的小鼠,其抗GBM肾炎肾脏损伤加重[13]。Aitman等[14]研究还发现特异性Fcgr3类似基因——Fcgr3相关基因序列(FCGR3B)的缺失,是引起Mφ过度活化和肾小球肾炎的一个决定性因素。在人类,FCGR3B(相当于大鼠定向进化同源的Fcgr3)的拷贝数量减少,与自身性免疫性疾病——系统性红斑狼疮并发的肾小球肾炎有关。脾酪氨酸激酶(Syk)是一种非受体酪氨酸激酶,是Fc和补体受体等细胞表面受体激活后的早期活化信号。Syk对于Mφ发挥Fc受体和补体诱导的吞噬及合成活性氧簇和细胞因子的功能至关重要。Syk抑制剂能够抑制肾脏Mφ浸润,减轻肾脏损伤,改善狼疮NZW/NZB小鼠的生存率[15]。尽管Sky抑制剂也能降低B细胞反应,减少肾小球免疫球蛋白沉积,但其抑制Mφ活化作用仍然发挥重要的保护效应。(4)其他促炎信号通路:与肾脏损伤相关的Mφ内促炎信号通路还有很多,如细胞因子信号通路、c-fms促炎信号通路,Janus激酶/信号传导子及转录激活子(JAK-STAT)信号通路及细胞因子信号通路抑制蛋白1(SOCS1)和SOCS3蛋白等,SOCS1和SOCS3蛋白可调控Mφ、树突状细胞及T细胞活化。
单核巨噬细胞的分化机制Mφ的亚型与肾脏炎症、损伤、修复及纤维化关系密切,所以研究调控Mφ分化的分子机制显得尤其重要。多种信号分子、转录因子、表观遗传机制及转录后调节等机制参与了Mφ的表型转化。IFNs和TLR通过IRF/STAT(STAT1)信号通路,使得Mo向M1表型分化,而白细胞介素4(IL-4)、IL-13可能通过JAK3/STAT6使其向M2表型分化。有研究显示,受损的近段上皮细胞可通过独立于IL-4、IL-13的途径诱导Mo/Mφ向M2型分化[16],近端小管分泌巨噬细胞集落刺激因子(CSF-1),介导Mφ向M2型分化[17-19]。Ⅰ型和Ⅱ型IL-4受体活化STAT6,STAT6可激活促进向M2型分化的转录因子。IL-10刺激STAT3相关的基因表达,进而促进Mo/Mφ向M2型分化。SOCS家族成员调控由STAT介导的Mφ活化。IL-4和IFN-γ上调SOCS1和SOCS3蛋白表达,而SOCS1和SOCS3能够分别抑制STAT1和STAT3的活化,从而抑制Mo/Mφ向M1分化。
核内受体过氧化物酶体增殖物激活受体γ(PPARγ)和PPARδ调控不同的基因,这些基因与M2型的活化和氧代谢有关。STAT6与PPARγ、Kruppe样因子(KLF4)具有协同作用[20]。KLF4与STAT6协同诱导M2型相关基因表达,同时由于缺乏炎性转录因子NF-κB辅助激活蛋白,从而抑制M1型相关基因表达。KLF2通过抑制NF-κB/低氧诱导因子1α(HIF-1α)[21]活性来调节Mφ的活化过程。NF-κB活化也能够刺激消炎相关基因的表达及肿瘤相关巨噬细胞(TAM)的分化。而且,研究表明p50 NF-κB同型二聚体是Mφ分化为M2型的基础[22]。HIF-1α和HIF-2α在M1型和M2型中表达不同。在M1型中,HIF-1α调节一氧化氮合酶2(NOS2)的合成;在M2型中,HIF-2α调节精氨酸酶1(Arg-1)的合成。
表观遗传改变和非编码RNAs也参与Mo/Mφ的分化[23]。在小鼠Mφ中,IL-4诱导组蛋白脱甲基酶JMJD3上调,JMJD3改变染色体修饰,从而促进M2型基因表达并且抑制M1型基因。一项研究结果表明,在人类Mφ中miRNA-155可降低IL-13Ra1亚基蛋白水平,从而减少M2型基因的表达[24]。
单核巨噬细胞的增生机制在人类肾脏疾病和小鼠模型中都观察到Mφ会发生增殖[25,26]。Mφ上的CSF-1的受体——c-fms活化促进自身增殖并且向M2型分化[27]。近段小管上皮细胞分泌CSF-1,促进Mφ增殖[17]。在怀孕的小鼠子宫中,也观察到CSF-1可介导Mφ的增殖和分化[28],组织炎症过程中,骨髓来源的炎性Mo和固有Mφ都会发生增殖。当组织炎症消散后,组织内固有Mφ对M-CSF反应敏感,继而增殖。此外,IL-4、IL-13促进Mφ增殖,并且IL-4不依赖于CSF-1R介导Mφ增殖。实验中观察到IL-4刺激组织内固有Mφ增殖聚集,而不依赖血液中的Mo募集至组织中[1]。除上述因子可刺激Mφ增殖外,IL-34、GM-CSF也能发挥类似效应[29]。
M-CSF通过Ras、Erk、蛋白激酶B(AKT)等信号通路影响核内转录因子Myc,AP-1和Ets调控Mφ增殖[30],其中Ap-1和Ets可分别活化促增殖因子c-myc和c-myb。MafB和c-Maf是Mφ增殖的核心调节子[31]。在Mφ分化过程中,MafB和c-Maf上调,并且诱导细胞周期阻滞。在缺失MafB和c-Maf时,成熟的Mo和Mφ可在不丢失表型和功能情况下发生增殖。其次,PI3K/AKT信号通路在IL-4诱导Mφ增殖中发挥重要作用,而且IL-4可通过活化STAT6抑制M-CSF诱导的增殖。
最近研究表明,腹腔Mφ增殖与转录因子GATA6有关,Mφ特异性敲除gata6后,增殖能力降低并且表型发生改变[32],而维甲酸可活化gata6基因,并且诱导腹腔Mφ功能性分化[33]。
Mφ亚型与肾脏疾病的关系
大量研究表明在不同类型肾脏病变中,Mφ是肾组织中浸润最多也是最重要的炎症细胞之一。
M1型Mφ在肾组织炎症反应及损伤过程中起了非常重要的作用。多种因素特别是病原相关分子模式(PAMPs),如细菌胞壁成分脂多糖、鞭毛素、富含胞嘧啶-鸟嘌呤(CpG)的微生物寡聚脱氧核苷酸等,能够在体外直接激活Mφ。而肾脏内外的感染、肾脏固有细胞的坏死等可使局部微环境中富集PAMPs,PAMPs通过结合包含TLR在内的多种模式识别受体(PRRs)高效激活Mφ内NF-κB和MAPK信号途径,产生多种促炎细胞因子(如TNF-α、IL-1β、IL-12、IL-18、IL-23及IL-6等),趋化因子[如巨噬细胞炎性蛋白1(MIP-1)、CXC趋化因子1(CXCL1)],活性氧簇和活性氮簇及其他炎性介质等[34]。此外,沉积于肾小球及与白细胞受体(包括激活免疫球蛋白Fc受体和补体受体)结合的免疫复合物,在特定情况下也能激活Mφ,产生与病原相关分子类似的作用[35]。在小鼠肾小球肾炎模型中,预先清除体内Mφ或中和介导Mφ浸润的趋化因子,可缓解蛋白尿和肾脏病变;再次输入骨髓来源的Mφ(BMDM)或NP8383细胞株(肺泡Mφ)则能够部分恢复肾组织炎症反应和蛋白尿水平[36]。此外,输入经IFNg处理的Mφ可加重肾脏损伤,而输入经类固醇处理的Mφ,或抑制Mφ中的JNK及NF-κB信号通路可缓解肾小球和肾小管间质损伤。同样,在肾缺血再灌注(I/R)损伤模型中,研究表明早期浸润的致炎M1型Mφ是肾小管间质损伤的关键致病因素[18]。
M2型Mφ参与了肾组织损伤后的修复过程。Wang等[11]输入M2型Mφ至慢性炎性肾脏病的小鼠体内,可减轻肾脏损伤、促进修复及下调浸润Mφ的炎性细胞因子和趋化因子的表达,说明M2型Mφ可促进慢性炎症的消散及损伤组织的修复。M2型Mφ通过胞吞作用清除炎症及损伤细胞和细胞碎片等,从而对肾组织起到保护作用。此外大量研究证实M2型Mφ还可通过以下多个方面参与肾组织损伤后的修复过程:分泌IL-4及IL-10来抑制M1型Mφ的分化;分泌营养因子,促进血管生成和伤口愈合;合成纤维连接蛋白1(FN-1)、转化生长因子β1(TGF-β1)及胰岛素样生长因子1(IGF-1)等介导组织修复和增生;产生精氨酸酶,减少致炎分子NO产生,进而抑制炎症反应[37];产生抗炎细胞因子,抑制效应淋巴细胞增生和作用[38],如M2a和M2c在体外均能抑制CD4+T细胞增生,IL-10、TGF-β或共刺激分子B7-H4的中和抗体能部分阻断该效应,而且M2a和M2c型Mφ也可抑制CD8+T细胞介导的小管细胞毒性作用[6]。M2c型Mφ可促进CD4+CD25-T细胞转变为Foxp3+细胞;此外,在炎症因素刺激下,骨髓起源的细胞可通过转分化途径,替代肾小管上皮细胞、系膜细胞及肾小球中的固有细胞,促进损伤后修复[39,40]。
然而,目前对于M2型Mφ在肾组织纤维化中的作用存在一些争议。一般认为M2型Mφ的持续浸润可通过以下几个方面促进肾组织纤维化:(1)产生Arg-1和IL-13,促进纤维化。由于精氨酸酶能将精氨酸水解为鸟氨酸,生成多胺谷氨酸和脯氨酸,而这些都是胶原合成必需的物质,因而Mφ来源的精氨酸酶能直接促进纤维化。另外,Mφ可分泌IL-13,IL-13可直接促进肌成纤维细胞产生胶原基质。(2)产生和分泌TGF-β[41]。Mφ是肾组织中TGF-β的主要来源之一,而肾小管上皮细胞是将TGF-β由无活性转变为活性状态的主要部位。αvβ6整联蛋白则介导了无活性TGF-β在上皮侧的活化。TGF-β不仅能激活并促使肌成纤维细胞增生,还能使上皮细胞在细胞周期中停滞和死亡,进而损伤肾组织。此外,TGF-β还可使肾小管上皮细胞转分化为肌成纤维母细胞。(3)Mφ可分化为一种纤维细胞,直接产生细胞外基质,促进纤维化。尽管研究证实了M2型Mφ在促进肾间质纤维化中的作用,但也有研究发现在阿霉素肾病模型中输入M2型Mφ可减少自身TGF-β分泌并具有抗炎和减轻纤维化的作用[11,42]。因此,M2型Mφ所发挥的作用不仅与肾脏疾病模型有关,而且也与肾脏病变的发展阶段有关。
小结:目前在多种肾脏病动物模型中,清除循环中Mo/Mφ、阻断介导Mo/Mφ浸润的各种趋化/细胞因子、基因遗传修饰Mφ及体外定向活化Mφ等,Mφ靶向治疗的疗效已经得到了证实。但是由于人体内环境及疾病状态错综复杂,Mφ真正成为治疗人类肾脏疾病的靶点还有很多难题需要解决。例如:如何辨别M2型在不同肾脏疾病中的亚型;如何在恰当的时机阻断M2型致纤维化作用,使其仅表现为介导上皮修复及再生的功能;如何明确Mφ与其他细胞类型(如T细胞、B细胞、树突状细胞及小管上皮细胞等)之间的相互作用等。随着基础研究的深入,我们对Mφ在肾脏炎症、损伤、修复及纤维化中的作用也会有更为深入的认识,这为将来在临床中开展Mφ靶向治疗奠定基础。
1Jenkins SJ,Ruckerl D,Cook PC,et al.Local macrophage proliferation,rather than recruitment from the blood,is a signature of TH2 inflammation.Science,2011,332(6035):1284-1288.
2Lu J,Cao Q,Zheng D,et al.Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease.Kidney Int,2013,84(4):745-755.
3Zhang JD,Patel MB,Griffiths R,et al.Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor-mediated kidney fibrosis.J Clin Invest,2014,124(5):2198-2203.
4Lech M,Gröbmayr R,Ryu M,et al.Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy.J Am Soc Nephrol,2014,25(2):292-304.
5Duffield JS.The inflammatory macrophage:a story of Jekyll and Hyde.Clin Sci (Lond),2003,104(1):27-38.
6Wang Y,Harris DC.Macrophages in renal disease.J Am Soc Nephrol,2011,22(1):21-27.
7Anders HJ,Ryu M.Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis.Kidney Int,2011,80(9):915-925.
8Flanc RS,Ma FY,Tesch GH,et al.A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis.Kidney Int,2007,72(6):698-708.
9Stambe C,Atkins RC,Tesch GH,et al.Blockade of p38alpha MAPK ameliorates acute inflammatory renal injury in rat anti-GBM glomerulonephritis.J Am Soc Nephrol,2003,14(2):338-351.
10 Wilson HM,Chettibi S,Jobin C,et al.Inhibition of macrophage nuclear factor-kappaB leads to a dominant anti-inflammatory phenotype that attenuates glomerular inflammation in vivo.Am J Pathol,2005,167(1):27-37.
11 Wang Y,Wang YP,Zheng G,et al.Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease.Kidney Int,2007,72(3):290-299.
12 Park SY,Ueda S,Ohno H,et al.Resistance of Fc receptor- deficient mice to fatal glomerulonephritis.J Clin Invest,1998,102(6):1229-1238.
13 Nakamura A,Yuasa T,Ujike A,et al.Fcgamma receptor IIB-deficient mice develop Goodpasture’s syndrome upon immunization with type IV collagen:a novel murine model for autoimmune glomerular basement membrane disease.J Exp Med,2000,191(5):899-906.
14 Aitman TJ,Dong R,Vyse TJ,et al.Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans.Nature,2006,439(7078):851-855.
15 Bahjat FR,Pine PR,Reitsma A,et al.An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus.Arthritis Rheum,2008,58(5):1433-1444.
16 Lee S,Huen S,Nishio H,et al.Distinct macrophage phenotypes contribute to kidney injury and repair.J Am Soc Nephrol,2011,22(2):317-326.
17 Zhang MZ,Yao B,Yang S,et al.CSF-1 signaling mediates recovery from acute kidney injury.J Clin Invest,2012,122(12):4519-4532.
18 Menke J,Iwata Y,Rabacal WA,et al.CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice.J Clin Invest,2009,119(8):2330-2342.
19 Taguchi K,Okada A,Kitamura H,et al.Colony-Stimulating Factor-1 Signaling Suppresses Renal Crystal Formation.J Am Soc Nephrol,2014.
20 Szanto A,Balint BL,Nagy ZS,et al.STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells.Immunity,2010,33(5):699-712.
21 Mahabeleshwar GH,Kawanami D,Sharma N,et al.The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock.Immunity,2011,34(5):715-728.
22 Porta C,Rimoldi M,Raes G,et al.Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB.Proc Natl Acad Sci USA,2009,106(35):14978-14983.
23 Satoh T,Takeuchi O,Vandenbon A,et al.The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection.Nat Immunol,2010,11(10):936-944.
24 Martinez-Nunez RT,Louafi F,Sanchez-Elsner T.The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1).J Biol Chem,2011,286(3):1786-1794.
25 Yang N,Isbel NM,Nikolic-Paterson DJ,et al.Local macrophage proliferation in human glomerulonephritis.Kidney Int,1998,54(1):143-151.
26 Schreiner GF,Cotran RS,Pardo V,et al.A mononuclear cell component in experimental immunological glomerulonephritis.J Exp Med,1978,147(2):369-384.
27 Zhang MZ,Yao B,Yang S,et al.CSF-1 signaling mediates recovery from acute kidney injury.J Clin Invest,2012,122(12):4519-4532.
28 Tagliani E,Shi C,Nancy P,et al.Coordinate regulation of tissue macrophage and dendritic cell population dynamics by CSF-1.J Exp Med,2011,208(9):1901-1916.
29 Sieweke MH,Allen JE.Beyond stem cells:self-renewal of differentiated macrophages.Science,2013,342(6161):1242974.
30 Smith JL,Schaffner AE,Hofmeister JK,et al.ets-2 is a target for an akt (Protein kinase B)/jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice.Mol Cell Biol,2000,20(21):8026-8034.
31 Aziz A,Soucie E,Sarrazin S,et al.MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages.Science,2009,326(5954):867-871.
32 Rosas M,Davies LC,Giles PJ,et al.The Transcription Factor Gata6 Links Tissue Macrophage Phenotype and Proliferative Renewal.Science,2014,344(6184):645-648.
33 Okabe Y,Medzhitov R.Tissue-Specific Signals Control Reversible Program of Localization and Functional Polarization of Macrophages.Cell,2014,157(4):832-844.
34 Medzhitov R.Approaching the asymptote:20 years later.Immunity,2009,30(6):766-775.
35 Ravetch JV,Bolland S.IgG Fc receptors.Annu Rev Immunol,2001,19:275-290.
36 Ikezumi Y,Hurst LA,Masaki T,et al.Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation.Kidney Int,2003,63(1):83-95.
37 Arnold L,Henry A,Poron F,et al.Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis.J Exp Med,2007,204(5):1057-1069.
38 Schebesch C,Kodelja V,Müller C,et al.Alternatively activated macrophages actively inhibit proliferation of peripheral blood lymphocytes and CD4+ T cells in vitro.Immunology,1997,92(4):478-486.
39 Poulsom R,Forbes SJ,Hodivala-Dilke K,et al.Bone marrow contributes to renal parenchymal turnover and regeneration.J Pathol,2001,195(2):229-235.
40 Kale S,Karihaloo A,Clark PR,et al.Bone marrow stem cells contribute to repair of the ischemically injured renal tubule.TJ Clin Invest,2003,112(1):42-49.
41 Kang HR,Cho SJ,Lee CG,et al.Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent,bid-activated pathway that involves matrix metalloproteinase-12.J Biol Chem,2007,282(10):7723-7732.
42 Cao Q,Wang Y,Zheng D,et al.IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis.J Am Soc Nephrol,2010,21(6):933-942.
