TNF-α 通过死亡受体途径促进IFN-γ 诱导NIT-1 细胞凋亡①
2014-03-18董世访李桂清江伟凡曹朝晖许桂莲
董世访 李桂清 江伟凡 杨 菲 曹朝晖 许桂莲
(第三军医大学基础部免疫学教研室,重庆 400038)
1 型糖尿病(Type 1 diabetes mellitus,T1DM)是一种以慢性炎症和胰岛β 细胞破坏为特征的自身免疫疾病[1]。T1DM 的发病常见于儿童或青少年,严重的并发症是导致儿童早期死亡的主要因素。T1DM 发病机制复杂,胰岛β 细胞死亡主要由细胞凋亡引起[2-4]。引起β 细胞凋亡的因素较多,其中自身反应性T 淋巴细胞、巨噬细胞入侵胰岛是主要的诱因。目前较多学者认为浸润的巨噬细胞、T 淋巴细胞释放的促炎症细胞因子如γ-干扰素(Interferon-γ,IFN-γ)、肿瘤坏死因子α (Tumor necrosis factor-α,TNF-α)、白介素1β(Interleukin-1β,IL-1β)连同Fas 配体(FasL)、穿孔素和颗粒酶B 是导致β 细胞凋亡、早期胰岛炎的形成及T1DM 发展的主要因素。而且,各种炎症细胞因子是通过联合而不是单独作用诱导了β 细胞凋亡,炎症细胞因子的组合方式和分布因动物模型而异[5-7]。因此,更加明确地了解胰岛β 细胞中不同细胞因子组合激活的凋亡信号通路,对于防治T1DM 实施个性化诊疗策略是十分必要的。
细胞凋亡信号通路主要包括死亡受体通路、内质网通路和内在的线粒体通路。已有研究表明凋亡的线粒体通路参与了IFN-γ 和TNF-α 联合诱导的β细胞凋亡[8-10],但死亡受体途径是否在β 细胞凋亡中发挥重要作用仍存在争议;死亡受体途径涉及IFN-γ 和TNF-α 诱导的NIT-1 细胞凋亡的文献报道较少。因此,本研究以小鼠胰岛β 细胞株NIT-1 为细胞模型,观察IFN-γ 和TNF-α 联合作用对NIT-1细胞凋亡的影响,并进一步研究了导致NIT-1 细胞凋亡的可能分子机制。
1 材料与方法
1.1 主要材料与试剂 小鼠胰岛β 细胞株NIT-1细胞购自上海汉博生物科技有限公司;rmIFN-γ 和rmTNF-α 购自美国Peprotech 公司;DMEM 培养基购自美国Gibco 公司;胎牛血清购自美国Hyclone 公司;Hoechst33258、MTT、DMSO 购自美国Sigma 公司;兔抗小鼠Caspase-3、cleaved Caspase-3 单克隆抗体购自美国CST 公司;兔抗鼠Caspase-8、多聚ADP-核糖聚合酶[poly(ADP-ribose)polymerase,PARP]单克隆抗体、β-actin 多克隆抗体、ECL 试剂盒购自碧云天公司;其余均为国产试剂(分析纯级)。
1.2 细胞培养 NIT-1 细胞用含10%胎牛血清的DMEM 培养基,在37℃、5%CO2、95%饱和湿度培养箱中传代培养,取对数生长期细胞进行实验。
1.3 MTT 分析 取对数生长期的细胞,以2 ×104个/孔密度接种于96 孔板,用IFN-γ 和TNF-α 单独或联合处理NIT-1 细胞24、48 和72 h。其中未加IFN-γ 和TNF-α 刺激的细胞(即0 h)为空白对照组。细胞因子处理后,每孔加20 μl MTT(5 g/L),于37℃、5%CO2培养箱中培养4 h 后,弃培养基,每孔加150 μl DMSO,待紫色结晶完全溶解后,用多功能酶标仪测定波长为490 nm 的吸光度值。设定未处理组(即0 h)细胞生长活力为100%,处理组细胞生长活力(%)=处理组吸光度值/未处理组吸光度值×100%。
1.4 细胞的形态学分析 细胞(1 ×104/孔)接种于96 孔板,IFN-γ 和TNF-α 联合刺激细胞24、48 和72 h,用倒置显微镜观察细胞形态变化。另一组实验中,细胞(3 × 105/孔)接种于6 孔板的盖玻片上,IFN-γ 和TNF-α 联合刺激细胞48 h。其中未加IFNγ 和TNF-α 刺激的细胞(即0 h)为空白对照组。收获细胞,PBS 洗3 遍后,用5 μg/ml Hoechst 33258 于室温下避光染色15 min。激光共聚焦显微镜观察细胞核形态变化。
1.5 Western blot 细胞(3 ×105/孔)接种6 孔板,IFN-γ 和TNF-α 联合处理细胞12 h 和24 h。其中未加IFN-γ 和TNF-α 刺激的细胞(即0 h)为空白对照组。收获细胞,预冷PBS 洗2 遍后,用细胞裂解液(150 mmol/L NaCl,5 mmol/L EDTA,1 mmol/L PMSF,1 % Triton X-100,1% 脱氧胆酸钠,0.1 %SDS)在4℃下裂解细胞提取细胞总蛋白,用Nano-Drop ND-1000 微型分光光度计测定蛋白质含量。每组蛋白样品取35 μg 进行SDS-PAGE 电泳分离,采用电转移中的湿转法将凝胶中的蛋白质转印至PVDF 膜上,用含5%脱脂牛奶的封闭液封闭2 h,用一定比例稀释的 Caspase-3、cleaved Caspase-3、Caspase-8、PARP 和β-actin 一抗室温孵育2 h 或4℃孵育过夜,再用辣根过氧化物酶标记的二抗室温孵育0.5~1 h,ECL 显色,胶片显影。以β-actin 为参照分析目的蛋白的相对含量。
1.6 统计学分析 采用GraphPad Prism 5.0 统计软件进行分析,数据以±s 表示,统计方法采用方差齐性分析和非配对的t 检验,以P <0.05 为差异有统计学意义。
2 结果
2.1 TNF-α 联合IFN-γ 抑制NIT-1 细胞的生长 MTT 结果表明,TNF-α 和IFN-γ 单独处理NIT-1 细胞48 h 和72 h,产生轻微的毒性作用,但两者结合显著地降低了细胞活力,且具有时间依赖性(表1)。
2.2 TNF-α 联合IFN-γ 对NIT-1 细胞形态的影响 倒置显微镜下观察发现,随着TNF-α 和IFN-γ 联合作用时间的延长,NIT-1 细胞形态逐渐由不规则瓦片状,开始变圆甚至漂浮,细胞密度明显降低,细胞之间空隙增加(图1)。Hoechst 33258 染色细胞核结果表明,未处理组细胞核染色质分布均匀、呈正常的低密度蓝染;而IFN-γ 和TNF-α 联合处理组细胞出现凋亡小体、核固缩、呈致密浓染(图2)。
2.3 TNF-α 联 合IFN-γ 处 理NIT-1 细 胞,诱 导Caspase-3,-8 的活化 Caspases 家族是细胞内执行凋亡程序的一类关键蛋白。如图3 所示,TNF-α 和IFNγ 联合处理细胞12 h 后,Caspase-3,-8 均出现明显的裂解片段,表明此二者在细胞因子的诱导下被激活。
2.4 PARP 的裂解 DNA 修复酶PARP 是Caspase-3 的一种作用底物蛋白,在细胞因子TNF-α 和IFNγ 的联合作用下被裂解失活(见图4)。
表1 TNF-α 联合IFN-γ 对NIT-1 细胞生长活力的影响(±s,n=4)Tab.1 Effect of TNF-α combined with IFN-γ on viability of NIT-1 cells (±s,n=4)

表1 TNF-α 联合IFN-γ 对NIT-1 细胞生长活力的影响(±s,n=4)Tab.1 Effect of TNF-α combined with IFN-γ on viability of NIT-1 cells (±s,n=4)
Note:1)P <0.01 compared with TNF-α group,2)P <0.01 compared with IFN-γ group.
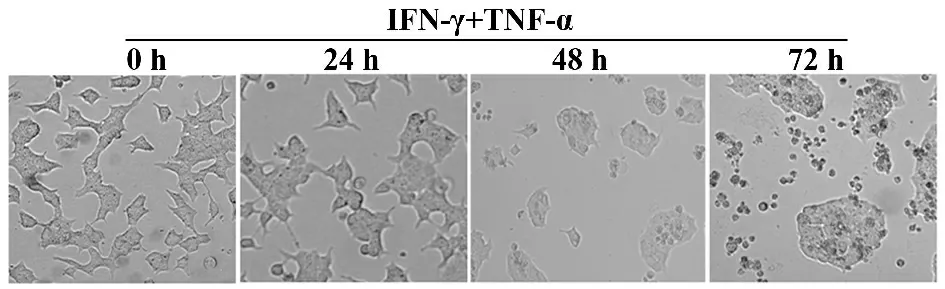
图1 TNF-α 联合IFN-γ 对NIT-1 细胞形态学的影响Fig.1 Effect of TNF-α and IFN-γ on morphological changes of NIT-1 cells (×200)
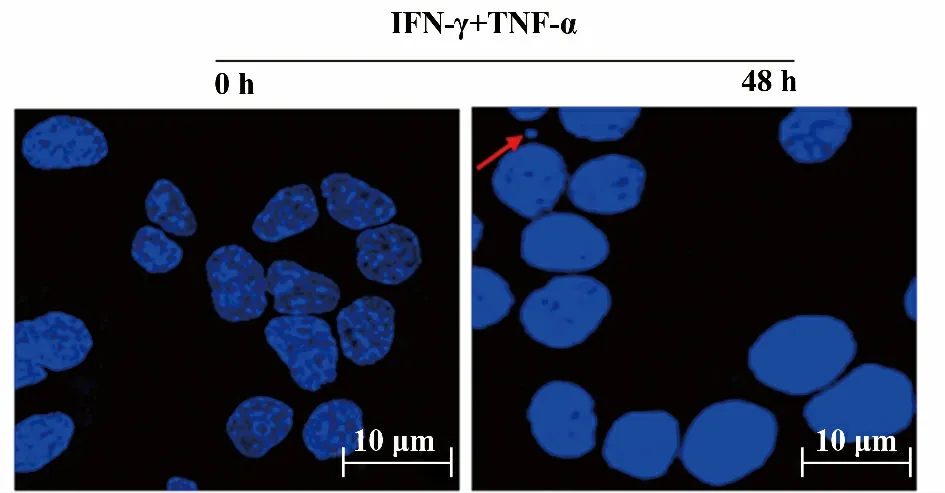
图2 TNF-α 联合IFN-γ 对NIT-1 细胞核形态的影响Fig.2 Effect of TNF-α and IFN-γ on nuclear changes of NIT-1 cells

图3 TNF-α 联合IFN-γ 对NIT-1 细胞Caspase-3,-8 活化的影响Fig.3 Effect of TNF-α and IFN-γ treatment on activation of Caspase-3,-8 in NIT-1 cells

图4 TNF-α 联合IFN-γ 对NIT-1 细胞PARP 活性的影响Fig.4 Effect of TNF-α and IFN-γ on activation of PARP in NIT-1 cells
3 讨论
T1DM 胰岛炎的发展过程中,不同人群炎症细胞因子的种类及它们之间的相互作用可能发生变化。细胞因子TNF-α 结合IFN-γ 诱导β 细胞死亡的确切分子机制还不是很清楚[5]。全面了解不同细胞因子组合在不同细胞模型中诱导的细胞凋亡信号途径,将有利于寻找个性化的药物靶标并为未来开展个性化治疗提供理论依据。
本实验以小鼠胰岛β 细胞株NIT-1 细胞为模型,体外研究IFN-γ 和TNF-α 协同作用诱导β 细胞凋亡及相关的信号途径。MTT 结果显示,TNF-α 和IFN-γ 联合作用能显著抑制NIT-1 细胞活力,且呈时间依赖性,而TNF-α、IFN-γ 单独处理对细胞仅产生轻微的毒性作用(表1)。随着TNF-α 和IFN-γ 作用于细胞的时间延长,细胞变圆甚至漂浮,细胞密度明显降低,细胞间隙增加(图1),细胞核出现凋亡小体、核固缩、呈致密浓染(图2)。提示IFN-γ 和TNF-α 对NIT-1 细胞的毒性作用是由于细胞凋亡所致,以上结果与Suk 等[11]在MIN6 细胞中观察的结果一致。IFN-γ 和TNF-α 引起NIT-1 细胞凋亡的分子机制是否涉及细胞凋亡的死亡受体途径?未见报道。
凋亡是一种自主的程序性细胞死亡形式,Caspases 家族蛋白在凋亡过程中起着关键作用[12-14]。信号传递激活外在的死亡受体途径,即经死亡受体相关的包含死亡结构域蛋白(Fas-Associated Death Domain-Containing Protein,FADD)信号传递激活Caspase-8。Caspase-3 是Caspase 级联反应中一种共同的下游效应分子,是凋亡的执行者。Caspase-8 通过裂解Caspase-3 的无活性酶原形式而激活Caspase-3。Caspase-3 再裂解细胞内其他蛋白底物如PARP,PARP 是DNA 修复酶,它被裂解后失去正常功能,激活受PARP 负调控的核酸内切酶活性,最终导致核小体DNA 断裂,从而触发细胞凋亡程序[15-17]。
本研究中,IFN-γ 和TNF-α 联合作用于小鼠胰岛β 细胞株NIT-1 细胞不同的时间后,提取蛋白质,进行Western blot 分析。结果显示,随着作用时间的增加,细胞内Caspase-8 和Caspase-3 相继被裂解激活(图3),引起了下游分子PARP 的裂解(图4)。我们的研究结果证实,IFN-γ 和TNF-α 联合处理诱导了NIT-1 细胞凋亡与其激活死亡受体进行信号转导有关。至于是否还涉及其他相关凋亡途径,还有待进一步研究。Fas 或Fas 相关的包含死亡结构域的蛋白可能作为药物靶点在防治T1DM 方面发挥一定作用。
[1]Kim HS,Lee MS.Role of innate immunity in triggering and tuning of autoimmune diabetes[J].Curr Mol Med,2009,9:30-44.
[2]Lightfoot YL,Chen J,Mathews CE.Role of the mitochondria in immune-mediated apoptotic death of the human pancreatic β cell line bLox5[J].Plos One,2011,6:e20617.
[3]Barthson J,Germano CM,Moore F,et al.Cytokines tumor necrosis factor-α and interferon-γ induce pancreatic β-cell apoptosis through STAT1-mediated Bim protein activation[J].J Biol Chem,2011,286:39632-39643.
[4]Gurzov EN,Germano CM,Cunha DA,et al.p53 Up-regulated modulator of apoptosis (PUMA)activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress [J].J Biol Chem,2010,285:19910-19920.
[5]Eizirik DL,Colli ML,Ortis F.The role of inflammation in insulitis and beta-cell loss in type 1 diabetes[J].Nature Reviews Endocrinology,2009,5:219-226.
[6]Grunnet LG,Aikin R,Tonnesen MF,et al.Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells[J].Diabetes,2009,58:1807-1815.
[7]McKenzie MD,Dudek NL,Mariana L,et al.Perforin and Fas induced by IFN-γ and TNF-α mediate beta cell death by OT-I CTL[J].Int Immunol,2006,18:837-846.
[8]Gurzov EN,Eizirik DL.Bcl-2 proteins in diabetes:Mitochondrial pathways of beta-cell death and dysfunction[J].Trends in Cell Biol,2011,21:424-431.
[9]Kim S,Kim HS,Chung KW,et al.Essential role for signal transducer and activator of transcription-1 in pancreatic b-cell death and autoimmune type 1 diabetes of nonobese diabetic mice[J].Diabetes,2007,56:2561-2568.
[10]Allagnat F,Cunha D,Moore F,et al.Mcl-1 downregulation by proinflammatory cytokines and palmitate is an early event contributing to β-cell apoptosis[J].Cell Death and Differ,2011,18:328-337.
[11]Suk K,Kim S,Kim YH,et al.IFN-γ/TNF-α synergism as the final effector in autoimmune diabetes:A key role for STAT1/IFN regulatory factor-1 pathway in pancreatic beta cell death[J].J Immunol,2001,166:4481-4489.
[12]Hengartner MO.The biochemistry of apoptosis[J].Nature,2000,407:770-776.
[13]Danial NN,Korsmeyer SJ.Cell death:critical control points[J].Cell,2004,116:205-219.
[14]Scorrano L,Korsmeyer S J.Mechanisms of cytochrome c release by proapoptotic BCL-2 family members[J].Biochem Biophys Res Commun,2003,304:437-444.
[15]Tibbetts MD,Zheng L,Lenardo MJ.The death effector domain protein family:regulators of cellular homeostasis[J].Nat Immunol,2003,4:404-409.
[16]Huang Q,Shen HM.To die or to live:the dual role of poly(ADP-ribose)polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage [J].Autophagy,2009,5:273-276.
[17]J Park S,KimJ A,Choi S,et al.Superoxide is a potential culprit of caspase-3 dependent endothelial cell death induced by lysophosphatidylcholine[J].Physiol Pharmacol,2010,61:375-385.
