遗传性脊髓小脑共济失调3 型一家系7 例
2014-03-11余叶菁孙占用贾艳丽王建华
余叶菁,孙占用,贾艳丽,王建华
脊髓小脑性共济失调(spinocerebellar ataxia,SCA)是遗传性共济失调的主要类型,成年期发病、常染色体显性遗传及共济失调是本病的共同特征。本病分为多种亚型,其中SCA3 型常见于我国[1],但其遗传特征、临床表现及确切的分子机制仍不完全明了。我们于2013 年6 月发现1 例经基因检测证实的SCA3 病例,现报道如下。
1 病例摘要
先证者(Ⅲ7)男,59 岁。因行走不稳10 余年,间断头晕、恶心7 y,加重3 y 于2013 年6 月来我院就诊。患者于10余年前出现行走不稳,似醉酒步态,无头晕,症状逐渐加重,当时查头部CT(2003 年6 月)提示小脑萎缩,未予重视。7 y前开始出现间断头晕,伴恶心、呕吐、饮水呛咳,无吞咽困难、饮水呛咳及听力下降等,头晕平卧位加重,坐位减轻,睁眼加重,闭眼减轻。不伴视物旋转、耳鸣、听力减退;不伴视力减退,肢体麻木等。症状持续1~2 d 自行缓解,每月发作3~4次,遂就诊我院行头部MRI(2007 年10 月7 日)检查提示小脑萎缩;颈椎MRI(2007 年10 月7 日)提示脑干、小脑萎缩。随后至广州空军医院就诊,考虑“多系统萎缩”可能性大,患者与家属未予重视,回家休养。3 y 前完全不能行走,饮水呛咳症状加重,出现言语不清。无视物旋转、耳鸣、肢体麻木、大小便功能障碍等。近半年来头晕症状加重,不能平卧,饮水呛咳严重,进食减少,体重下降,经休息头晕症状持续不能缓解。既往30 y 前开始反复出现气胸,每次经闭式胸腔引流治疗后好转,于7 y 前发现肺大泡,并行肺大泡切除术。个人史:足月顺产,生后躯体及智能发育正常,吸烟30 y,约40支/日,已戒烟7 y,饮酒30 y,约白酒4 两/日,已戒酒7 y,无冶游及性病史。查体:言语含糊,吟诗样语言,软腭上提对称,悬雍垂居中,咽反射减弱。记忆力、计算力、定向力均未见异常,粗侧视力未见异常,眼球各方向运动自如,可引出水平眼震,右侧凝视时明显,四肢肌张力减低,四肢肌力均为Ⅳ级,双侧轮替试验不灵活,双侧指鼻试验、跟膝胫试验不稳,Romberg 征睁闭眼均阳性,感觉系统未见异常,双下肢音叉振动觉减退,四肢腱反射对称活跃,双侧Hoffmann 征阴性,双侧Babinski 征及Chaddock 征未引出,颈无抵抗,Kernig 征阴性。心肺腹检查未见异常,头部、四肢及躯干未见畸形。辅助检查:头部核磁共振提示小脑半球和蚓部脑沟增宽,脑干周围脑池扩大,大脑各脑叶皮质无萎缩;6 y 后复查提示小脑萎缩较前稍有加重(见图1)。基因检测结果:遗传性脊髓小脑性共济失调3 型(SCA3)三核苷酸(CAG)n 重复大于50。
家系调查(见图2):四代共7 例患者(男6 例女1 例),分别为先证者的伯父(Ⅱ1)、姑姑(Ⅱ2)、父亲(Ⅱ3)、大哥(Ⅲ1)、二哥(Ⅲ5)及侄子(Ⅳ3)。第Ⅱ代平均发病年龄55岁,第Ⅲ代平均发病年龄55 岁,第Ⅳ代发病年龄34 岁。除患者本人外,第2 代和第3 代发病者均已去世。全部病例的主要症状为走路不稳、吞咽困难、言语含糊及头晕等。
2 讨论
脊髓小脑性共济失调(SCA)是遗传性共济失调的主要类型,主要包括SCA1~SCA21。成年期发病、常染色体显性遗传及共济失调等是本病的共同特征,并表现为连续数代中发病年龄提前和病情加重的特征(遗传早现)。根据是否伴眼肌麻痹、锥体外系症状及视网膜色素变性等SCA 可分为3型:常染色体显性遗传小脑共济失调(autosomal dominant cerebellar ataxias,ADCA)Ⅰ型、Ⅱ型和Ⅲ型,SCA1、SCA2、SCA3、SCA4、SCA8、SCA12、SCA13、SCA17、SCA19、SCA21 均属于ADCA Ⅰ型。其中SCA3 常见于中国,又名Machado-Joseph病(Machado-Joseph disease,MJD),具有眼震、进行性共济失调、构音障碍、锥体系及锥体外系征、以及肌肉萎缩等临床表现[1]。临床可以分成3 型:Ⅰ型发病年龄早且重复次数多;Ⅱ、Ⅲ型发病年龄晚,三核苷酸CAG 重复次数相对少。通常所指的为Ⅰ型即MJD1,是遗传性共济失调最常见的亚型,在中国约占全部SCA 的48.23%[2],致病基因位于14 号染色体长臂,已知染色体14q24.3-32.1 内的MJD1 3’端的CAG三核苷酸拷贝数异常扩增形成多聚谷氨酸(polyglutamine,PolyQ)链,导致蛋白代谢异常,引起细胞凋亡而致病[3]。CAG 拷贝数与患者发病年龄呈负相关,即重复扩增越多,发病年龄越早,病情进展越迅速[4]。CAG 拷贝数的大小可对发病年龄的早晚作出45%~60%的解释[5],拷贝数与临床症状同样相关,拷贝数高于73 次者常出现锥体束征,而低于73次者多表现周围神经病变。中间等位基因在传代过程中有扩增到病理范围的趋势,从而导致新发病例的出现。据报道,拷贝数在42~60 次也可致病[6,7],提示中间等位基因的扩增可能是一种完全外显性的突变。
本家系为SCA3 临床分型的Ⅱ型或Ⅲ型,平均发病年龄50 岁左右,男女比例为6∶1。家族患者主要表现为进行性小脑共济失调、构音障碍、饮水呛咳、头晕及锥体束征等,各代之间遗传早现现象不典型,男女均有发病,符合常染色体显性遗传模式。本例患者基因检查显示三核苷酸(CAG)n重复大于50,根据相关报道[6,7],可以诊断SCA3。遗憾的是我们仅收集了先证者本人的血样进行了基因检测结果,家系中其他成员并未行基因检测。总之,尽管目前对SCA3 的诊断有了很大进步,但确切分子机制仍未阐明,还有待于深入研究。
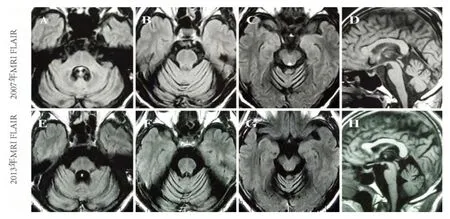
图1 头部MRI:A~D 小脑半球和蚓部脑沟增宽,脑干周围脑池扩大;E~H 6 y 后复查显示小脑萎缩较前稍有加重
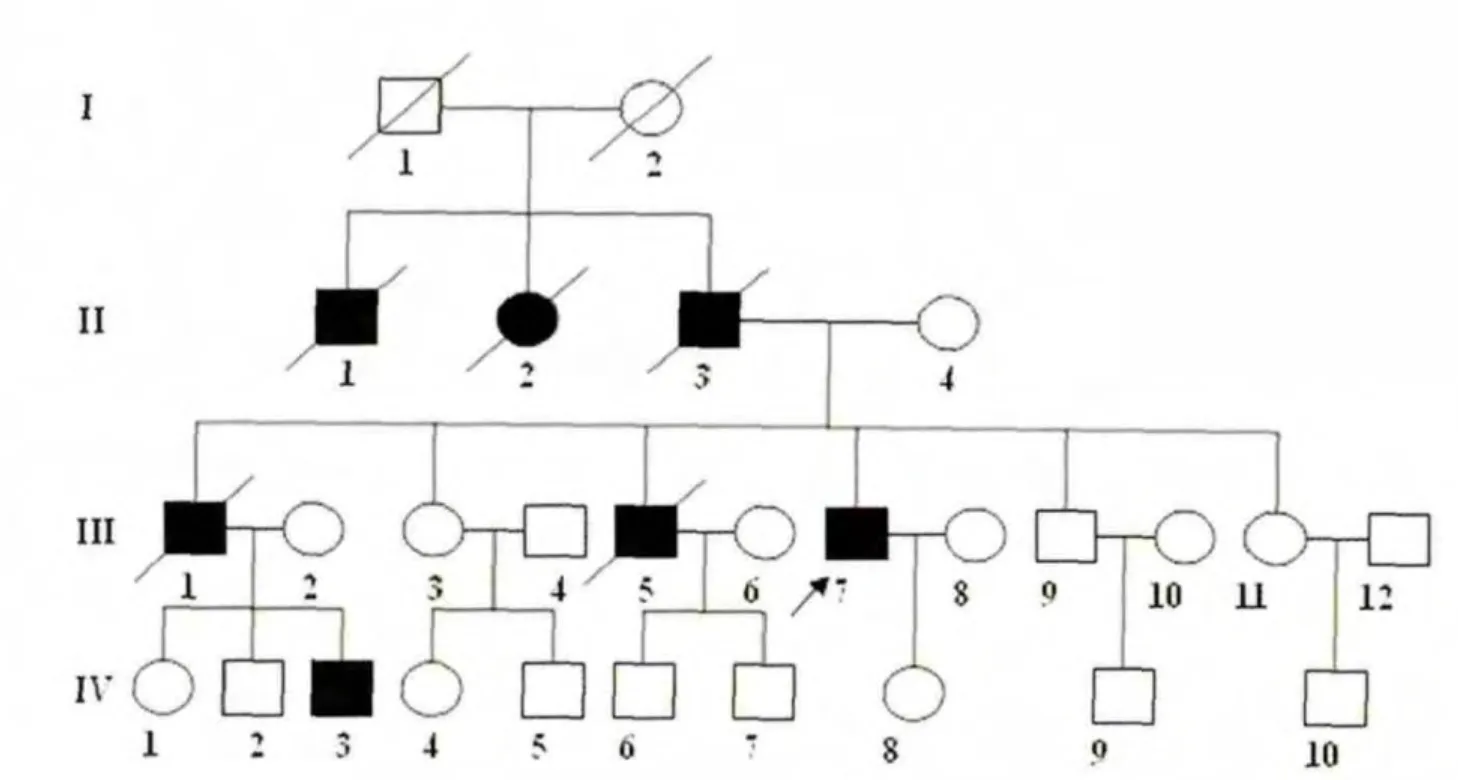
图2 SCA3 患者家系图
[1]Ashizawa T,Figueroa KP,Perlman SL,et al.Clinical characteristics of patients with spinocerebellar ataxias 1,2,3 and 6 in the US;a prospective observational study[J].Orphanet J Rare Dis,2013,13(8):177-185.
[2]Tang B,Liu C,Shen L,et al.Frequency of SCA1,SCA2,SCA3/MJD,SCA6,SCA7,and DRPIA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds[J].Arch Neurol,2000,57(4):540-544.
[3]Takazaki KA,D'Abreu A,Nucci A,et al.Dysautonomia is frequent in Machado-Joseph disease:clinical and neurophysiological evaluation[J].Cerebellum,2013,12(4):513-519.
[4]Evers MM,Tran HD,Zalachoras I,et al.Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3:removal of the CAG containing exon[J].Neurobiol Dis,2013,58:49-56.
[5]van de Warrenburg BP,Sinke RJ,Verschuuren-Bemelmans CC,et al.Spinocerebellar ataxias in the Netherlands:prevalence and age at onset variance analysis[J].Neurology,2002,58(5):702-708.
[6]Gu W,Ma H,Wang K,et al.The shortest expanded allele of the MJD1 gene in a Chinese MJD kindred with autonomic dysfunction[J].Eur Neurol,2004,52(2):107-111.
[7]van Alfen N,Sinke RJ,Zwarts MJ,et al.Intermediate CAG repeat lengths(53,54)for SCA3/MJD are associated with an abnormal phenotype[J].Ann Neurol,2001,49(6):805-807.
