鬼臼毒素类新药的研发思路
2014-02-16许晓辉孙陶利许莉莉朱玉婷
许晓辉,孙陶利,许莉莉,朱玉婷
鬼臼毒素类新药的研发思路
许晓辉,孙陶利,许莉莉,朱玉婷
鬼臼毒素是有一定药效活性的环木脂内酯,具有抗肿瘤、抗病毒、抗风湿、杀虫等活性。其中,抗肿瘤活性最引人关注,常常作为研究开发抗肿瘤药物的先导化合物。但鬼臼毒素水溶性差、易产生多药耐药性和毒性较强等缺点限制了它的临床应用,因此通过结构修饰寻找高效低毒的鬼臼毒素衍生物是鬼臼毒素类化合物研究的重点。本文介绍鬼臼毒素衍生物的药效团模型、构效关系模型以及当前对鬼臼毒素进行结构修饰的研究思路。
鬼臼毒素;药效团;定量构效关系
鬼臼毒素(图1-1)是一种存在于小檗科的八角莲属、鬼臼属和山荷叶属等多种植物中具有一定药效活性的环木脂内酯化合物。在我国,蕴含鬼臼毒素类的植物多集中于青海、甘肃、西藏、四川等地[1-4]。研究发现,鬼臼毒素类衍生物能够抑制免疫、抗炎以及治疗疟疾、牛皮癣和重症肌无力等症。同时,鬼臼毒素能够使肿瘤细胞周期停留在G2/M期,抑制细胞中期的有丝分裂,对小细胞肺癌、睾丸癌、白细胞癌、淋巴肉瘤、神经胶质瘤、霍奇金淋巴瘤等多种肿瘤有特殊疗效[5-9]。但是鬼臼毒素母体化合物存在一些缺点使它的临床应用受到限制,比如水溶性差、不良反应多、易产生耐药性,为了得到活性优良的鬼臼毒素类药物,研究者以鬼臼毒素母体化合物为先导化合物对鬼臼毒素进行结构修饰改造,合成了众多的鬼臼毒素类衍生物,通过对这些衍生物的抗肿瘤活性进行筛选,得到了一些相对母体鬼臼毒素活性有很大改善的药物。
1 临床上应用的鬼臼毒素类抗肿瘤药物
目前,临床上应用的鬼臼毒素类抗肿瘤药物主要有依托泊苷(图1-2)、替尼泊苷(图1-3)及依托泊苷磷酸盐(图1-4)等,后续开发的具有潜在抗肿瘤活性并有望成为新药的有GL-311(图1-5)、F 11782(图1-6)、F14512(图1-7)、TOP-53(图1-8)、NPF(图1-9)等。其中,GL-311克服了多种肿瘤细胞对依托泊苷的耐药性,对多耐药性的癌细胞也有很好的活性;F 11782是一种含有全氟苯酚结构的表鬼臼毒素衍生物,现处于临床Ⅱ期试验中[10-11];F14512是拓扑异构酶Ⅱ(topoisomeraseⅡ,TopoⅡ)抑制剂,于2009年9月进入了临床Ⅰ期试验[12-13];TOP-53、NPF已经进入临床Ⅱ期试验。
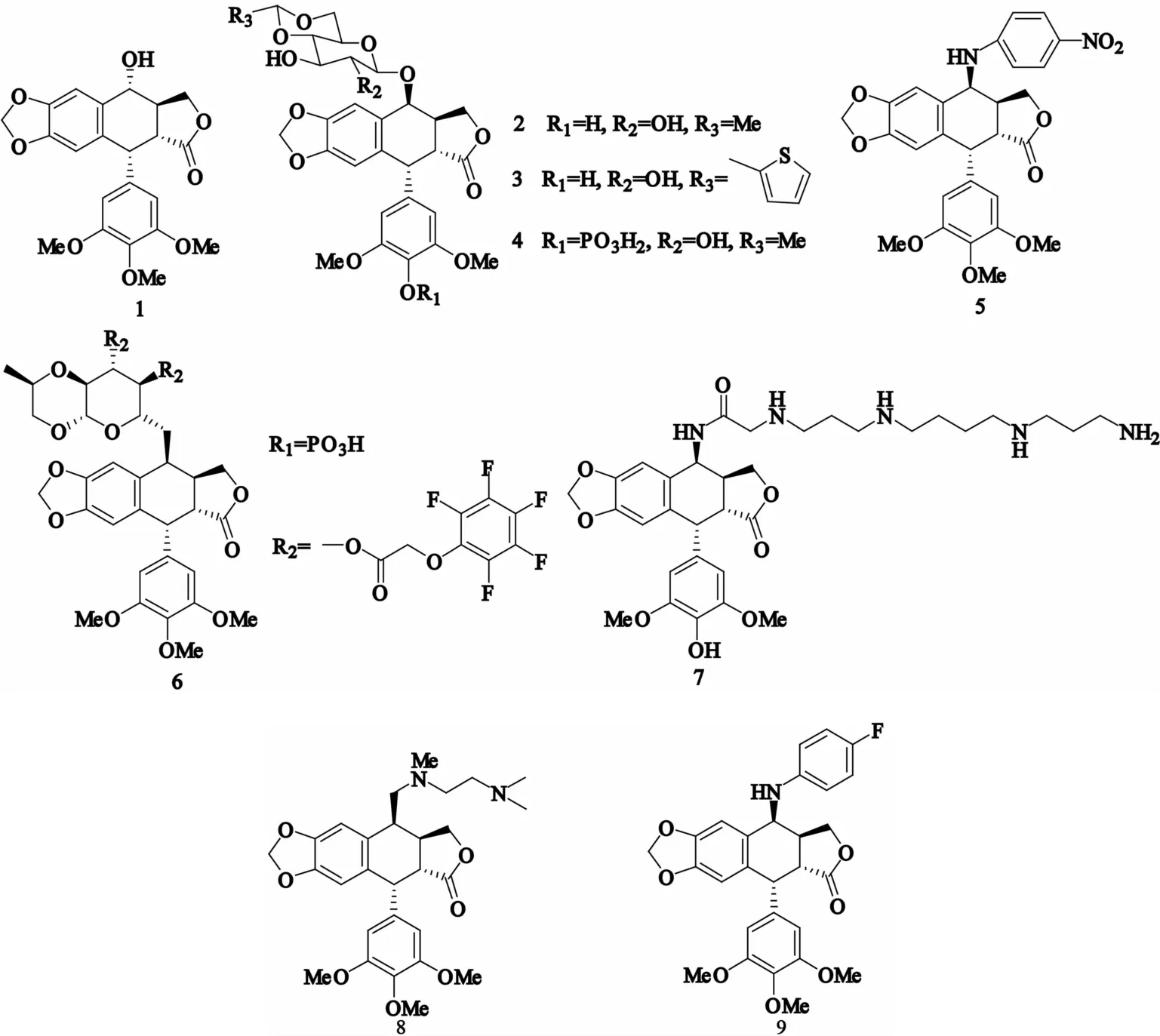
图1 鬼臼毒素类抗肿瘤药物的结构
2 鬼臼毒素类抗肿瘤药物的作用机制
人们对鬼臼毒素的抗肿瘤作用进行了研究,在构效关系、作用机制方面取得了一些进展,并已经深入到分子水平。鬼臼毒素类衍生物能够与微管蛋白结合,导致微管不能聚合,进而导致染色体在复制以后不能分离,促使细胞分裂停止在G2/M期,诱发细胞凋亡。鬼臼毒素衍生物还能抑制细胞对胸腺嘧啶T、尿嘧啶U、腺嘌呤A、鸟嘌呤G等核苷酸的摄取,阻碍细胞DNA、RNA以及蛋白质的合成,引起细胞凋亡。鬼臼毒素类衍生物能够捕获TopoⅡ引起的DNA断裂复合物,形成DNA-TopoⅡ-Drug三重稳定复合物,导致DNA不能正常重组,激活致死性蛋白酶的表达。研究发现,在生物体内,依托泊苷E环酚羟基在体内通过生物氧化反应生成一种半醌酮式的自由基中间体。这种半醌酮式的自由基中间体能够产生自由基与DNA形成配合物致使DNA断裂而使DNA失活。同时,半醌酮式的自由基还能与细胞色素P450形成邻苯二酚衍生物,邻苯二酚衍生物进一步氧化成邻苯二醌衍生物。这种邻苯二酚和邻苯二醌的衍生物形成自由基或以醌的形式与小牛胸腺DNA进行结合,引起DNA断裂,其作用机制见图2[14]。

图2 依托泊苷的邻苯醌自由基作用机制
3 鬼臼毒素结构修饰的理论依据
鬼臼毒素类化合物的抗肿瘤机制主要有2种,一是抑制微管组装,二是抑制TopoⅡ的活性。TopoⅡ是鬼臼毒素类化合物作用的靶点[15],通过对大量鬼臼毒素类衍生物构效关系的研究表明,C-4′位保留甲氧基的鬼臼毒素衍生物主要作用微管而抑制细胞的有丝分裂,使细胞周期停留在G2/M期,阻止微管蛋白形成微管;C-4′保留羟基的鬼臼毒素衍生物则是通过与TopoⅡ相互作用,使细胞周期停留在G期。此外,具有邻醌特性的鬼臼毒素衍生物,能导致自由基的形成,引起细胞DNA断裂。MacDonold等[16]通过分子模拟研究对嵌入式和非嵌入式的TopoⅡ抑制剂提出了一种通用的模式。该模式指出鬼臼毒素衍生物若要具有良好的DNA-TopoⅡ抑制活性必须具备3个药效团结构,即能与DNA最小凹槽键合点作用部分、DNA的插入部分、能够容纳不同结构的取代基的分子区域,其模型见图3[16]。该模型为药物化学家对鬼臼毒素母体进行修饰提供了理论依据,为进一步寻找鬼臼毒素类抗肿瘤药物具有重要指导意义。
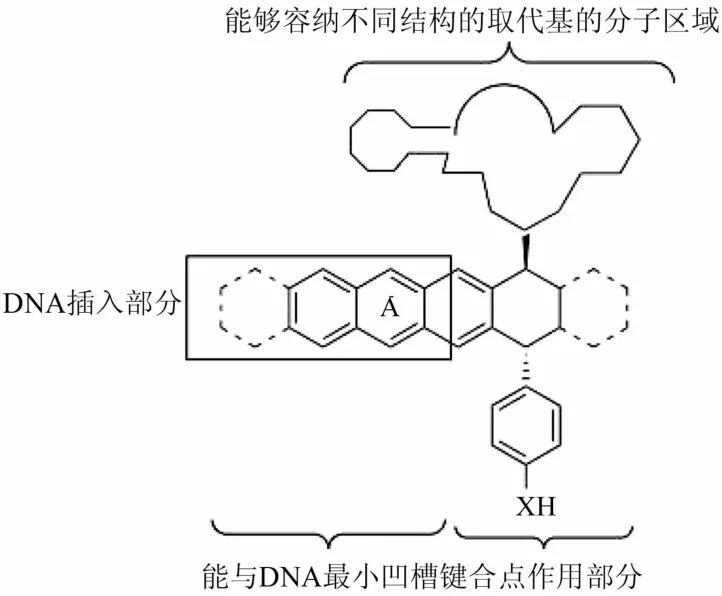
图3 鬼臼毒素衍生物对DNA-TopoⅡ抑制活性的药效团模型
4 鬼臼毒素衍生物的构效关系模型
鬼臼毒素类先导化合物在结构上都是2,3-丁内酯-1-芳基四氢萘的母核,共存在5个环,其中A、B、C、D 4个环形成的平面刚性结构,是DNA分子嵌入的重要功能区。为了寻找更加高效低毒的衍生物,研究者在尽可能保持其刚性结构的前提下,进行了大量的结构修饰工作。通过对上百个化合物运用构效关系、定量构效关系以及三维构效关系技术对其构效关系进行深入研究,结果表明鬼臼毒素类化合物要保持良好的抗肿瘤活性必须具备以下结构:①鬼臼毒素C-4位取代基保持4β构型;②C-4′位游离的酚羟基;③反式的内酯环结构;④鬼臼毒素A环中的二氧亚甲基;⑤可以自由旋转的E环。其结构的A、B、C、D、E环的分子骨架药效结构可概括在图4中[17]。

图4 鬼臼毒素及衍生物构效关系模型
5 鬼臼毒素结构修饰的研究思路
对鬼臼毒素的结构修饰位置有母核A、C、D、E环,主要集中于C环的C-4位和E环的C-4′位。鬼臼毒素C-4位的修饰有很多策略,主要有在C-4位的羟基通过酯连接其他小分子化合物,有通过对氧原子进行电子等排体(CH2、S、NH、Se)取代修饰的,有在C-4位胺基化成杂环并连接其他小分子化合物进行修饰的。近年来的修饰主要集中在C-4位胺的取代以及在C-4或者C-4′偶联其他抗肿瘤药物,形成拼合物的修饰策略。目前,已经合成的拼合物有与偏端霉素的拼合物[18]、与喜树碱的拼合物[19]、与紫杉醇的拼合物[20]、与嘧啶的拼合物[21-22]、与秋水仙碱的拼合物[23]、与长春瑞滨的拼合物[24]、与聚合物的拼合物[25]、与非甾体类消炎药的拼合物[26]等。Liu等[27-29]以不同的链接基合成了一系列顺铂-鬼臼毒素类拼合物、氟尿嘧啶-鬼臼毒素类拼合物,氮氧自由基-鬼臼毒素类拼合物,活性筛选发现、拼合物都表现出比原有母体高的抗肿瘤活性。
除了对鬼臼毒素母体结构从C-4位进行结构修饰和优化之外,也对其从C-4′位进行了大量的修饰工作。研究发现,C-4′位酚羟基若被烷基、酰基取代,生物活性出现下降;但是当封闭的C-4′酚羟基能够在体内游离暴露出来,有利于其氧化生成邻醌类衍生物,抗肿瘤活性增强。因此,C-4′位的修饰首先要利于C-4′-鬼臼毒素衍生物的C-4′酚羟基与靶标相互作用时,能够彻底暴露出来。C-4′进行修饰工作时,在C-4′位形成容易断裂的酯键、酰胺键为常见的修饰策略。
6 展望
目前,随着研究者对鬼臼毒素类药物抗肿瘤机制研究不断深入,构效关系不断揭示,为鬼臼毒素类新药从结构修饰上到药理活性筛选上都指明了一定的方向。以已经揭明的构效关系模型,通过合理的修饰设计,改变母体化合物的空间药效结构,改善母体化合物的低溶解性、化学稳定性,增加目标化合物在体内的渗透性、生物利用度,并以鬼臼毒素类衍生物的作用机制为依据设计合适的药理实验,筛选出具有潜在活性的药物。这些为进一步寻找高效低毒、结构新颖且利于与靶标结合的前体药物打好基础,为进一步研发鬼臼毒素类新药提供指导。
[1]Hartwell JL,Schrecker AW.The chemistry of Podophyllum[J].Fortschr Chem Org Naturst,1958,15:83-166.
[2]Stähelin H,von Wartburg A.From podophyllotoxin glucoside to etoposide[J].Prog Drug Res,1989,33:169-267.
[3]Canetta R,Hilgard P,Florentine S,et al.Current development of podophyllotoxins[J].Cancer Chemother Pharmacol,1982,7(2/3):93-98.
[4]陈毓亨.我国鬼臼类植物资源的研究[J].药学学报,1979,14(2):101-107.
[5]杨显志,邵华,张玲琪,等.鬼臼毒素资源研究现状[J].中草药,2001,32(11):1042-1044.
[6]刘海军,徐艳,苏国庆,等.桃儿七的研究进展[J].中草药,2004,35(1):98-100.
[7]Macrae WD,Neil Towers GH.Biological activities of lignans[J].Phytochemistry,1984,23(6):1207-1220.
[8]Canel C,Moraes RM,Dayan FE,et al.Podophyllotoxin[J].Phytochemistry,2000,54(2):115-120.
[9]Bohlin L,Rosen B.Podophyllotoxin derivatives:drug discovery and development[J].Drug Discov Today,1996,1(8):343-351.
[10]Perrin D,van Hille B,Barret JM,et al.F 11782,a novel epipodophylloid non-intercalating dual catalytic inhibitor of topoisomerasesⅠandⅡwith an originalmechanism of action[J].Biochem Pharmacol,2000,59(7):807-819.
[11]Barret JM,Etiévant C,Baudouin C,et al.F 11782,a novel catalytic inhibitor of topoisomerasesⅠandⅡ,induces atypical,yet cytotoxic DNA double-strand breaks in CHOK1 cells[J].Anticancer Res,2002,22(1A):187-192.
[12]Barret JM,Kruczynski A,VispéS,et al.F14512,a potent antitumor agent targeting topoisomeraseⅡvectored into cancer cells via the polyamine transportsystem[J].Cancer Res,2008,68(23):9845-9853.
[13]Kruczynski A,Vandenberghe I,Pillon A,et al.Preclinical activity of F14512,designed to target tumors expressing an active polyamine transport system[J].Invest New Drugs,2011,29(1):9-21.
[14]张甲强.稳定氮氧自由基自旋标记的鬼臼类化合物的合成及抗癌、抗氧化活性研究[D].兰州:兰州大学,2010.
[15]Gordaliza M,Castro MA,del Corral JM,et al.Antitumor properties of podophyllotoxin and related compounds[J]. Curr Pharm Des,2000,6(18):1811-1839.
[16]MacDonald TL,Lehnert EK,Loper JT,etal.On themechanism of interaction of DNA topoisomeraseⅡwith chemotherapeutic agents[M]//Potmesil M,Kohn KW.In DNA topoisomerase in cancer.New York:Oxford University Press,1991:119-214.
[17]Pilippe M,Emmanuel B,Thierry I,et al.Synthetic approaches to condensed aromatic analogues from etoposide.synthesis of a-ring pyridazine picroetoposide[J].Tetrahedron,1999,55:12805-12818.
[18]Ji Z,Wang HK,Bastow KF,et al.Antitumor agents.1771. Design,synthesis,and biological evaluation of novel etoposide analogs bearing pyrrolecarboxamiding group as DNA topoisomeraseⅡinhibitors[J].Bioorg Med Chem Lett,1997,7(5):607-612.
[19]Bastow KF,Wang HK,Cheng YC,et al.Antitumor agents--CLXXIII.Synthesis and evaluation of camptothecin-4 betaamino-4′-O-demethyl epipodophyllotoxin conjugates as inhibitors of mammalian DNA topoisomerases and as cytotoxic agents[J].Bioorg Med Chem,1997,5(8):1481-1488.
[20]Shi Q,Wang HK,Bastow KF,et al.Antitumor agents 210. Synthesis and evaluation of taxoid-epipodophyllotoxin conjugates as novel cytotoxic agents[J].Bioorg Med Chem,2001,9(11):2999-3004.
[21]Zhang FM,Yao XJ,Tian X,et al.Synthesis and biological evaluation of new 4beta-5-Fu-substituted 4′-demethylepipodophyllotoxin derivatives[J].Molecules,2006,11(11):849-857.
[22]Chen SW,Wang YH,Jin Y,etal.Synthesis and anti-HIV-1 activities of novel podophyllotoxin derivatives[J].Bioorg Med Chem Lett,2007,17(7):2091-2095.
[23]Passarella D,Peretto B,Blascoy Yepes R,et al.Synthesis and biological evaluation of novel thiocolchicine-podophyllotoxin conjugates[J].Eur JMed Chem,2010,45(1):219-226.
[24]Passarella D,Giardini A,Peretto B,et al.Inhibitors of tubulin polymerization:synthesis and biological evaluation of hybrids of vindoline,anhydrovinblastine and vinorelbine with thiocolchicine,podophyllotoxin and baccatinⅢ[J]. Bioorg Med Chem,2008,16(11):6269-6285.
[25]Lee NJ,Jeong IC,Cho MY,et al.Synthesis and in vitro antitumor activity of phthalimide polymers containing podophyllotoxin[J].Eur Polym J,2006,42:3352-3359.
[26]Uddin MJ,Smithson DC,Brown KM,et al.Podophyllotoxin analogues active versus Trypanosoma brucei[J].Bioorg Med Chem Lett,2010,20(5):1787-1791.
[27]Liu X,Zhang LL,Xu XH,et al.Synthesis and anticancer activity of dichloroplatinum(Ⅱ)complexes of podophyllotoxin[J].Bioorg Med Chem Lett,2013,23(13):3780-3784.
[28]Huang WT,Liu J,Liu JF,et al.Synthesis and biological evaluation of conjugates of deoxypodophyllotoxin and 5-FU as inducer of caspase-3 and-7[J].Eur J Med Chem,2012,49:48-54.
[29]Zhang ZW,Zhang JQ,Hui L,et al.First synthesis and biological evaluation of novel spin-labeled derivatives of deoxypodophyllotoxin[J].Eur JMed Chem,2010,45(4):1673-1677.
Study on the development of podophyllotoxin related new drugs XU Xiaohui1,SUN Taoli2,XU Lili1,ZHU Yuting3
(1.Pingliang Centre for Drug Inspection,Pingliang Gansu 744000,China;
2.The Basical Department of Medicine,Xiangnan University,Chenzhou Hunan 423000,China;3.People′s Hospital of Hanzhong,Hanzhong Shanxi723000,China)
Podophyllotoxin is cyclolignolide thathas some pharmacological activities,and it is known to have various functions and bioactivities such as antitumor,antivirus,anti-rheumatism and insecticide.Among them,the antitumor activity of podophyllotoxin is widely noted,so that it is taken as a lead compound in drug discovery of anticancer.However,there are some drawbacks,for example poor water solubility,easy to producemultidrug resistance and strong toxicity,these all limit its clinical application.So the focus of research is to find efficient and low toxicity of podophyllotoxin derivatives through the structuremodification.This paper introduces the pharmacophore model and quantitative structure activity relationships(QSAR)model of podophyllotoxin,and the research approach about its structuremodification.
Podophyllotoxins;Pharmacophore;Quantitative structure activity relationships(QSAR)
R914.5
A
2095-3097(2014)03-0162-04
10.3969/j.issn.2095-3097.2014.03.010
2014-02-23 本文编辑:张在文)
744000甘肃平凉,平凉市药品检验检测中心(许晓辉,许莉莉);423000湖南郴州,湘南学院基础医学部(孙陶利);723000陕西汉中,汉中市人民医院(朱玉婷)
