雷公藤多苷提取物中主要有效部位的含量比较*
2014-02-06石森林张茹萍范永升王新昌
何 昱 石森林 张茹萍 范永升 王新昌
(浙江中医药大学,浙江 杭州 310053)
雷公藤多苷是中药雷公藤的去皮根经提取分离得到的活性组分混合物。雷公藤多苷的生理活性由二萜类、三萜类、生物碱类等多种成分协同产生[1],但这些雷公藤多苷的有效成分也是其毒性成分[2-3],引发了临床上的众多不良反应[4-5]。由于目前国内对雷公藤多苷提取物及制剂仍缺乏较为完善的质量控制标准,为进一步合理应用雷公藤多苷,本研究应用紫外可见分光光度法对不同厂家的雷公藤多苷提取物中总二萜、总三萜、总生物碱3个有效部位进行了含量测定,以期为临床应用雷公藤多苷的安全有效提供实验依据。
1 材 料
METTLER AL104电子天平(梅特勒-托利多仪器有限公司),UV-2450型紫外可见分光光度计 (岛津,江苏);雷公藤甲素(中国药品生物制品检定所,批号111567-200502)、雷公藤红素(贵州迪大科技有限责任公司,批号A0106)、雷公藤次碱(浙江省农业科学院植物保护与微生物研究所,批号201012),层析用中性氧化铝(100-200目,上海五四化学试剂有限公司,批号100311),其他所用试剂均为分析纯。雷公藤多苷提取物(西安天本生物工程有限公司,批号TBT111106、TBT110412、TBT110801、TBT110624、TBT111026、TBC111201、TBC110927;湖南协力药业有限公司,批号20101209、20101108;桂林市三棱生物制品有限公司,批号 100808)。
2 方法与结果
2.1 总二萜的含量测定[6]
2.1.1 溶液制备 对照品溶液的制备:精密称取雷公藤甲素对照品5.0mg,加甲醇溶解并定容于5mL的容量瓶中,得质量浓度为1.0mg/mL的对照品溶液。供试品溶液的制备:精密称取雷公藤多苷提取物0.15 g,用无水乙醇溶解并稀释至25mL容量瓶中,即得。
2.1.2 检测波长的选择 分别精密吸取对照品溶液及供试品溶液各2.0mL,冰水浴中冷却后加入2%3,5-二硝基苯甲酸溶液 2.0 mL和 2mol/L KOH溶液 2.0 mL,冰水浴中放置10min后,自来水冲淋2min。以无水乙醇加显色剂为空白,在400~800 nm波长范围内进行扫描,结果对照品与供试品在544 nm处均有最大吸收,故选择544 nm作为雷公藤多苷样品中总二萜的检测波长(图 1)。
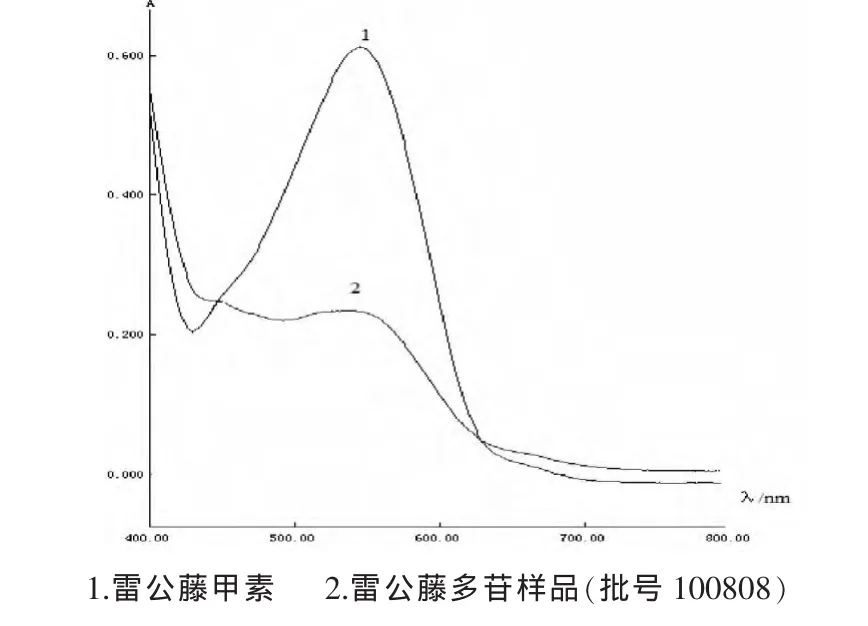
图1 雷公藤多苷中总二萜的可见吸收光谱图
2.1.3 标准曲线的绘制 分别精密移取雷公藤甲素对照品溶液 0.1、0.2、0.3、0.4、0.5、0.6mL 置于 10mL 容量瓶中,用无水乙醇稀释至刻度,吸取5.0mL稀释后的溶液于10mL容量瓶中,加入显色剂,定容,在544 nm下测定吸光度。以质量浓度(C)为横坐标,吸光度(A)为纵坐标,建立相应的回归方程为 A=20.289C+0.129,r=0.9998。
2.1.4 精密度试验 取质量浓度为 0.05mg/mL的雷公藤甲素对照品溶液,按“2.1.2 项”下方法显色后,连续测定6次吸光度值,其RSD=0.36%。
2.1.5 稳定性试验 批号20101209的雷公藤多苷样品按“2.1.1项”下制备供试品溶液,显色后每隔 5min测定吸光度值。结果35min内8次测得的吸光度RSD=2.88%,40min 内 9 次测得的 RSD=3.22%, 表明样品溶液在显色后35min内较为稳定,可以进行含量分析。
2.1.6 重复性试验 平行制备6份批号20101209的雷公藤多苷供试品溶液,依法测定后计算含量,结果6份供试品溶液中总二萜含量的RSD=2.01%。
2.1.7 加样回收率试验 精密称取 6份 0.075 g已知含量的雷公藤多苷样品(批号20101209),分别加入相应量的雷公藤甲素对照品,按“2.1.2项”下方法制备样品溶液,依法测定,计算得加样回收率为 (100.48±2.41)%,RSD=2.40%。
2.1.8 样品含量测定 各批次的雷公藤多苷提取物依法制备供试品溶液,显色后在544 nm下测定吸光度值,计算其中总二萜的含量,结果如表1所示。
2.2 总三萜的含量测定[7-8]
2.2.1 溶液的制备 对照品溶液的制备 精密称取雷公藤红素对照品5.0mg,加甲醇溶解并定容于5mL容量瓶中,配制成质量浓度为1.0mg/mL的对照品溶液。供试品溶液的制备:精密称取雷公藤多苷提取物0.1 g,置25mL容量瓶中,用甲醇溶解并定容,即得。
表1 不同厂家雷公藤多苷提取物中3个有效部位含量(mg,s)
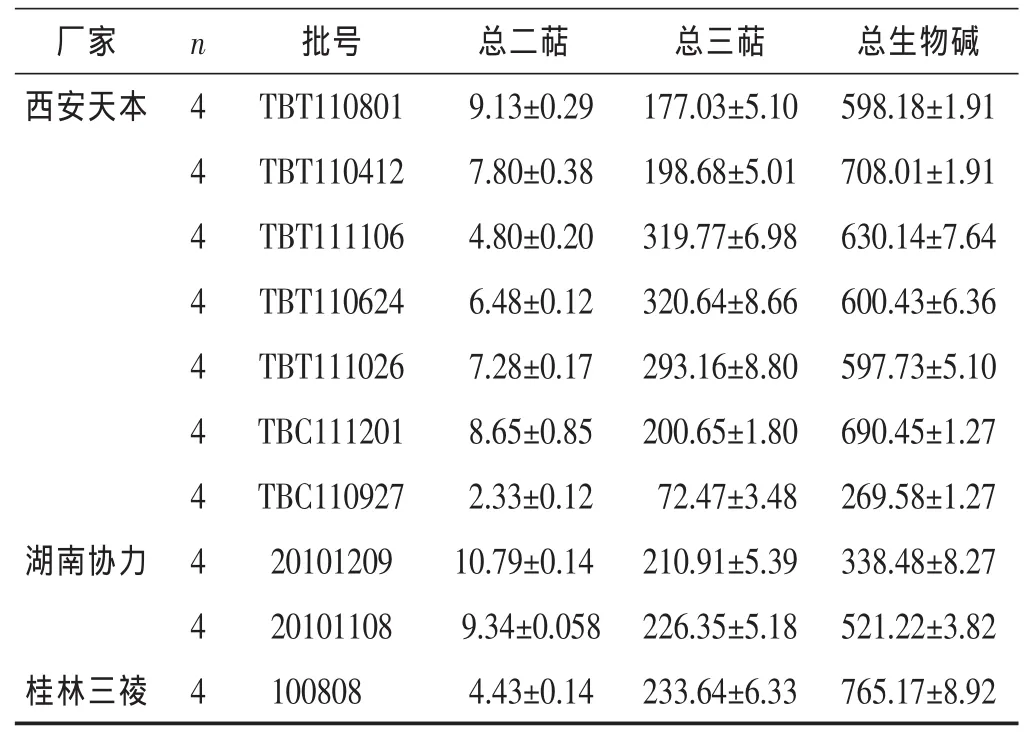
表1 不同厂家雷公藤多苷提取物中3个有效部位含量(mg,s)
厂家 n西安天本 4 4批号 总二萜 总三萜 总生物碱TBT110801 9.13±0.29 177.03±5.10 598.18±1.91 TBT110412 7.80±0.38 198.68±5.01 708.01±1.91 4 TBT111106 4.80±0.20 319.77±6.98 630.14±7.64 4 TBT110624 6.48±0.12 320.64±8.66 600.43±6.36 4 TBT111026 7.28±0.17 293.16±8.80 597.73±5.10 4 TBC111201 8.65±0.85 200.65±1.80 690.45±1.27 4 TBC110927 2.33±0.12 72.47±3.48 269.58±1.27湖南协力 4 20101209 10.79±0.14 210.91±5.39 338.48±8.27 4 20101108 9.34±0.058 226.35±5.18 521.22±3.82桂林三祾 4 100808 4.43±0.14 233.64±6.33 765.17±8.92
2.2.2 检测波长的选择 分别精密移取对照品溶液和供试品溶液各 0.02mL,100℃水浴挥干, 加入 0.4mL 5%香草醛-冰醋酸溶液和1.0mL高氯酸,摇匀后70℃水浴中加热45min,自来水冷却5min后再加入5.0mL冰醋酸,摇匀,10 min后,以冰醋酸为空白,在 400~600 nm波长范围内进行扫描,结果对照品溶液与供试品溶液在541 nm处均呈现最大吸收,在此波长下,显色剂对测定没有干扰(图2)。

图2 雷公藤多苷中总三萜的可见吸收光谱图
2.2.3 线性关系考察 精密移取雷公藤红素对照品溶液 0.1、0.2、0.3、0.4、0.5、0.6mL, 用甲醇稀释并定容于10mL 容量瓶中。再精密移取 2.5mL,按“2.2.2 项下”显色后测定541 nm处的吸光度值。以质量浓度(C)为横坐标,吸光度(A)为纵坐标,得回归方程A=25.183C+0.129,r=0.9998。
2.2.4 精密度试验 取1.0mg/mL的雷公藤红素对照品溶液,显色后连续测定6次吸光度值,其RSD=0.30%。
2.2.5 稳定性试验 取批号 TBT111026的雷公藤多苷供试品溶液在显色后1 h内测定吸光度。结果40min内7次测定的吸光度值RSD=2.48%,50min内8次测定的RSD=3.22%,表明在显色后40min内样品稳定。
2.2.6 重复性试验 取批号TBT111026的雷公藤多苷提取物,按“2.2.1项”下方法平行制备6份供试品溶液,依法测定,按标准曲线计算含量,结果6份供试品溶液中总三萜含量的RSD=1.92%。
2.2.7 加样回收率试验 取6份0.05g已知含量的雷公藤多苷样品(批号TBT111026),分别加入相应量的雷公藤红素对照品,按“2.2.1项”下制备供试品溶液,依法显色后测定, 得加样回收率为 (99.30±2.39)%,RSD=2.41%。
2.2.8 样品含量测定 各批次的雷公藤多苷提取物样品依法显色后在541 nm下测定吸光度,计算所得量减去总二萜的含量即为雷公藤多苷提取物中总三萜的含量,结果如表1所示。
2.3 总生物碱的含量测定[9]
2.3.1 溶液制备 对照品溶液的制备:精密称取雷公藤次碱对照品5.0mg,加乙腈溶解并定容于5mL容量瓶中,配制成浓度为1.0mg/mL的对照品溶液。供试品溶液的制备:精密称取雷公藤多苷样品0.15 g,置25 mL容量瓶中,加氯仿溶解并定容。精密移取2.0mL溶液,上3.5 g的中性氧化铝层析柱,以60mL氯仿∶甲醇(97∶3)洗脱,洗脱液水浴挥干,残渣用甲醇复溶后定容至50mL的容量瓶中。
2.3.2 检测波长的选择 分别将对照品溶液和供试品溶液在200~800 nm波长范围内进行扫描,结果对照品与供试品在225 nm、267 nm处均有最大吸收(图3)。为避免225 nm处雷公藤二萜类及三萜类的较强吸收干扰,故最终选择267 nm作为总生物碱的测定波长。

图3 雷公藤多苷中总生物碱的紫外吸收光谱图
2.3.3 线性关系考察 精密吸取雷公藤次碱对照品溶液 0.5、0.6、0.7、0.8、0.9、1.0mL 置 10mL 容量瓶, 用甲醇稀释后定容,测定267 nm处吸光度。以质量浓度(C)为横坐标,吸光度(A)为纵坐标,计算得回归方程A=4.629C+0.0305,r=0.9996。
2.3.4 精密度试验 取质量浓度为 0.05mg/mL的雷公藤次碱对照品溶液,连续测定6次吸光度,其RSD=0.041%。
2.3.5 稳定性试验 取批号20101108的雷公藤多苷供试品溶液, 分别于制备好后的 0、2、4、6、8、10、24 h,测定267 nm处的吸光度,结果24 h内RSD为0.23%,表明供试液在制备后24 h内稳定性良好。
2.3.6 重复性实验 取批号20101108的雷公藤多苷6 份,按“2.3.1 项”下方法平行制备 6 份供试品溶液,依法测定,计算得含量的RSD=2.19%。
2.3.7 加样回收率试验 取 6份 0.075g已知含量的雷公藤多苷提取物(批号20101108),分别加入相应量的雷公藤次碱对照品,依法测定,按标准曲线计算含量,得加样回收率为(100.32±2.22)%,RSD=2.21%。
2.3.8 样品含量测定 各批次的雷公藤多苷提取物按“2.3.1 项”下制备供试品溶液,依法测定,根据回归方程计算得雷公藤多苷提取物中总生物碱的含量,如表1所示。
3 讨 论
3.1 检测指标的确定 课题组在以往研究中已对雷公藤多苷提取物中的雷公藤甲素、雷公藤内酯酮、雷公藤晋碱、雷公藤次碱、雷公藤红素、雷公藤内酯甲等单体组分进行了HPLC含量测定[10]。但是,雷公藤多苷中众多的化学成分均有生物活性和毒性的报道,光凭单一成分并不能完全代表其药效、毒性等作用[11],因此本研究应用紫外可见分光光度法对雷公藤多苷中总二萜、总三萜及总生物碱3个有效部位进行了含量测定,以更全面地反映雷公藤多苷的化学组成情况。
3.2 定量结果的比较 从各批次雷公藤多苷提取物的测定结果来看,3个有效部位中,总二萜的含量最低,其次为总三萜,总生物碱的含量最高。同时,在3个厂家的各批次样品间,有效部位的最高含量与最低含量之间分别相差 4.6 倍、4.4 倍和 2.6 倍。雷公藤多苷作为治疗指数较小的药物[12],其组成成分含量上的显著差异值得引起相关生产厂家的重视。
[1] 郭雪红.雷公藤多苷的药理作用及临床应用概述[J].中成药,2010,32(7):1199-1202.
[2] 郭建龙,刘利民,江振洲,等.原料雷公藤多苷的化学成分研究[J].现代中药研究与实践,2011,25(4):41-44.
[3] 舒薇,刘继勇,杨帝顺.雷公藤多苷纳米乳的制备及体外透皮特性研究[J].中国药学杂志,2011,46(21):1651-1655.
[4] 杨臣,吴春敏.雷公藤多苷片的不良反应与防治[J].海峡药学,2013,25(6):276-277.
[5] 周炎仪,黄捷仪,李静玲,等.雷公藤制剂的用药安全性分析[J].临床合理用药杂志,2013,5(23):120.
[6] 张茹萍,何昱,石森林,等.雷公藤药材中6种有效成分以及总二萜内酯、总生物碱、总三萜的含量测定[J].中华中医药杂志,2013,28(1):224-229.
[7] 李宏杨,刘飞,张凤琴,等.6种冬青科苦丁茶总三萜类物质含量的比较研究[J].热带生物学报,2010,1(1):17-20.
[8] 王慧,沈鹏飞,曾荣今.枸骨根中三萜类物质含量的测定[J].南华大学学报:自然科学版,2011,25(4):105-108.
[9] 张洁,段宏泉.紫外分光光度法测定雷公藤多苷片和不同产地药材中总生物碱的含量[J].天津医科大学学报,2009,15(3):353-356.
[10]何昱,石森林,张茹萍,等.雷公藤多苷主要有效成分的含量研究[J].药物分析杂志,2013,33(2):197-200.
[11] Wong KF,Yuan Y,Luk JM.Tripterygium wilfordii bioactive compounds as anticancer and anti-inflammatory agents[J].Clinical and Experimental Pharmacology and Physiology,2012,(39):311-320.
[12] 张月琴.雷公藤多苷抗炎作用的研究[J].医药导报,2012,31(3):295-297.
