HPLC法同时测定补中益气丸中3种活性成分的含量
2014-01-24薛薇毛菊华王志芳
薛薇,毛菊华,王志芳
(1.广西中医药大学附属瑞康医院药学部,南宁 530011;2.丽水市食品药品检验所,浙江丽水 323000;3.广州市香雪制药股份有限公司,广州 510663)
HPLC法同时测定补中益气丸中3种活性成分的含量
薛薇1*,毛菊华2,王志芳3
(1.广西中医药大学附属瑞康医院药学部,南宁 530011;2.丽水市食品药品检验所,浙江丽水 323000;3.广州市香雪制药股份有限公司,广州 510663)
目的:建立同时测定补中益气丸中橙皮苷、毛蕊异黄酮苷、甘草酸含量的方法。方法:采用高相液相色谱法。色谱柱为Kromasil C18,流动相为甲醇-0.1%磷酸溶液(梯度洗脱),流速为1.0ml/min,柱温为30℃,检测波长为260nm,进样量为10μl。结果:橙皮苷、毛蕊异黄酮苷、甘草酸的检测质量浓度分别在0.0615~1.250、0.0255~0.510、0.0506~1.012mg/ml范围内与各自峰面积积分值呈良好的线性关系(r=0.9997、0.9998、0.9999);精密度、稳定性、重复性试验的RSD≤0.98%;平均加样回收率分别为99.19%、99.42%、101.13%,RSD分别为2.05%、2.43%、2.16%(n=9)。结论:该方法准确、可靠、简便易行,可用于补中益气丸的质量控制。
高效液相色谱法;补中益气丸;橙皮苷;毛蕊异黄酮苷;甘草酸
补中益气丸为常用补中益气药,原方为金元时期著名医家李东垣在其《脾胃论》中论述的“补中益气汤”方,方中黄芪为君药,党参、白术、甘草(炙)为臣药,当归、陈皮为佐药;升麻、柴胡为使药。现代研究表明,补中益气汤具有抗溃疡、调节免疫、抗胃黏膜损伤及调节激素等功效[1-4]。2010年版《中国药典》[5]中关于补中益气丸的质量控制仅为测定黄芪甲苷,但补中益气丸中有效成分较多,有文献报道,补中益气丸方中橙皮苷、毛蕊异黄酮苷和甘草酸均为有效成分[6-7]。因此,仅以检测黄芪甲苷作为衡量其质量的标准,具有一定的片面性,从而导致不同厂家生产的补中益气丸质量不一。目前,国内文献报道仅分别对补中益气丸中黄芪甲苷、橙皮甘、甘草酸的含量进行测定[8-9],并未对多个指标建立同时测定的方法。鉴于中药复方制剂化学成分的复杂性,为保证药品质量,笔者尝试采用高效液相色谱(HLPC)法同时测定补中益气丸中3种主要活性成分的含量。
1 材料
Summit P680HPLC仪,包括Summit P680A低压四元泵、DIONEX-UVD170U紫外检测器、ASI-100自动进样器、Chromelon色谱工作站(美国戴安公司);KQ-500型超声波清洗器(昆山市超声仪器有限公司);HH-S6数显恒温水浴锅(常州普天仪器制造有限公司);XS225A型分析天平(德国Sartorius公司)。
橙皮苷对照品(成都植标化纯生物技术有限公司,批号:120316);毛蕊异黄酮苷、甘草酸对照品(中国食品药品检定研究院,批号:130111、121124);补中益气丸(A公司,批号:201211027、201304034;B公司,批号:12082027;C公司,批号:121108;D公司,批号:N00014;E公司,批号:11D229;F公司,批号:130412);甲醇(色谱纯,德国默克公司);水(纯净水,华润怡宝食品饮料有限公司);其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Kromasil C18(250mm×4.6mm,5μm);流动相:流动相A为甲醇,流动相B为0.1%磷酸溶液,采用梯度洗脱(洗脱程序见表1);流速:1.0ml/min;柱温:25℃;检测波长:260nm;进样量:10μl。
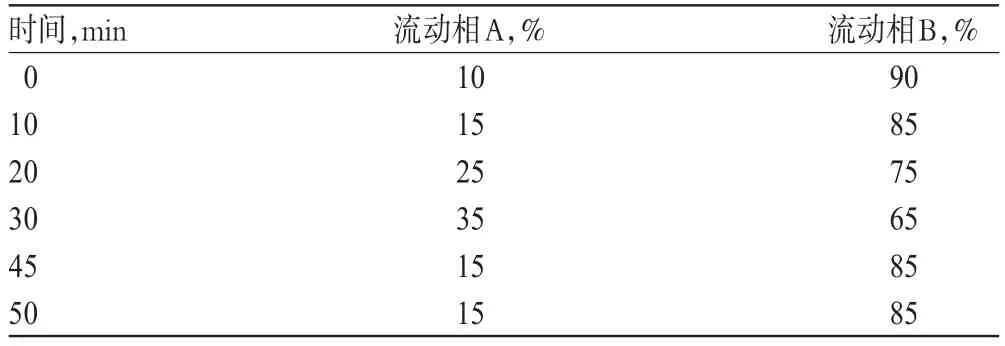
表1 流动相梯度洗脱程序Tab1 Gradient elution of mobile phase
2.2 溶液的制备
2.2.1 供试品溶液的制备 称取补中益气丸,切碎,取约1g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,密塞,称定质量,超声处理60min(功率:500W,频率:40kHz),放冷,称定质量,用甲醇补足减失的质量,用0.45µm孔径微孔滤膜滤过,取续滤液,即得。
2.2.2 对照品溶液的制备 精密称取橙皮苷对照品、毛蕊异黄酮苷对照品、甘草酸对照品适量,置于同一10ml棕色量瓶中,加入甲醇溶解并稀至刻度,摇匀,即得3种成分质量浓度分别为0.510、1.250、1.012mg/ml的对照品溶液。
2.2.3 空白溶液的制备 按补中益气丸处方比例制成不含黄芪、陈皮、甘草(炙)的空白对照品,按“2.2.1”项下供试品溶液的制备方法制备空白溶液。
2.3 系统适用性试验
精密吸取“2.2”项下供试品溶液、对照品溶液、空白溶液各10μl,注入HPLC仪,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。在该色谱条件下,3种成分可达到基线分离。理论板数按橙皮苷峰计算应不低于5000。

图1 高效液相色谱图A.空白;B.对照品;C.供试品1.毛蕊异黄酮苷;2.橙皮苷;3.甘草酸Fig1 HPLC chromatogramsA.blank;B.substance control;C.test samples;1.calycosin glycoside;2.hesperidin;3.glycyrrhizic
2.4 线性关系考察
分别精密吸取“2.2.2”项下对照品溶液1、0.75、0.5、0.2、0.1、0.05ml,置于1ml量瓶中,分别以甲醇定容至刻度,摇匀,得到不同质量浓度的系列溶液,按“2.1”项下色谱条件进样测定。以检测质量浓度(x)为横坐标,峰面积(y)为纵坐标,进行线性回归,回归方程及线性范围见表2。

表2 回归方程及线性范围Tab2 Results of regression equation and linear range of 3components
2.5 精密度试验
取“2.2.2”项下对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次。结果,橙皮苷、毛蕊异黄酮苷、甘草酸的RSD分别为0.24%、0.98%、0.79%,表明仪器的精密度良好。
2.6 稳定性试验
取“2.2.1”项下供试品溶液(批号:201211027)适量,分别于放置0、4、8、12、16、20、24h时按“2.1”项下色谱条件进样测定。结果,橙皮苷、毛蕊异黄酮苷、甘草酸的RSD分别为0.91%、0.26%、0.67%,表明供试品溶液在24h内稳定性良好。
2.7 重复性试验
取样品(批号:201211027)内容物适量,按“2.2.1”项下方法制备供试品溶液,共6份,按“2.1”项下色谱条件进样测定。结果,橙皮苷、毛蕊异黄酮苷、甘草酸的RSD分别为0.49%、0.67%、0.98%,表明本方法的重复性良好。
2.8 加样回收率试验
取已知含量的样品(批号:201211027)内容物约0.2g,共9份,分别加入一定量的橙皮苷、毛蕊异黄酮苷、甘草酸对照品溶液,按“2.2.1”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定并计算加样回收率,结果见表3。
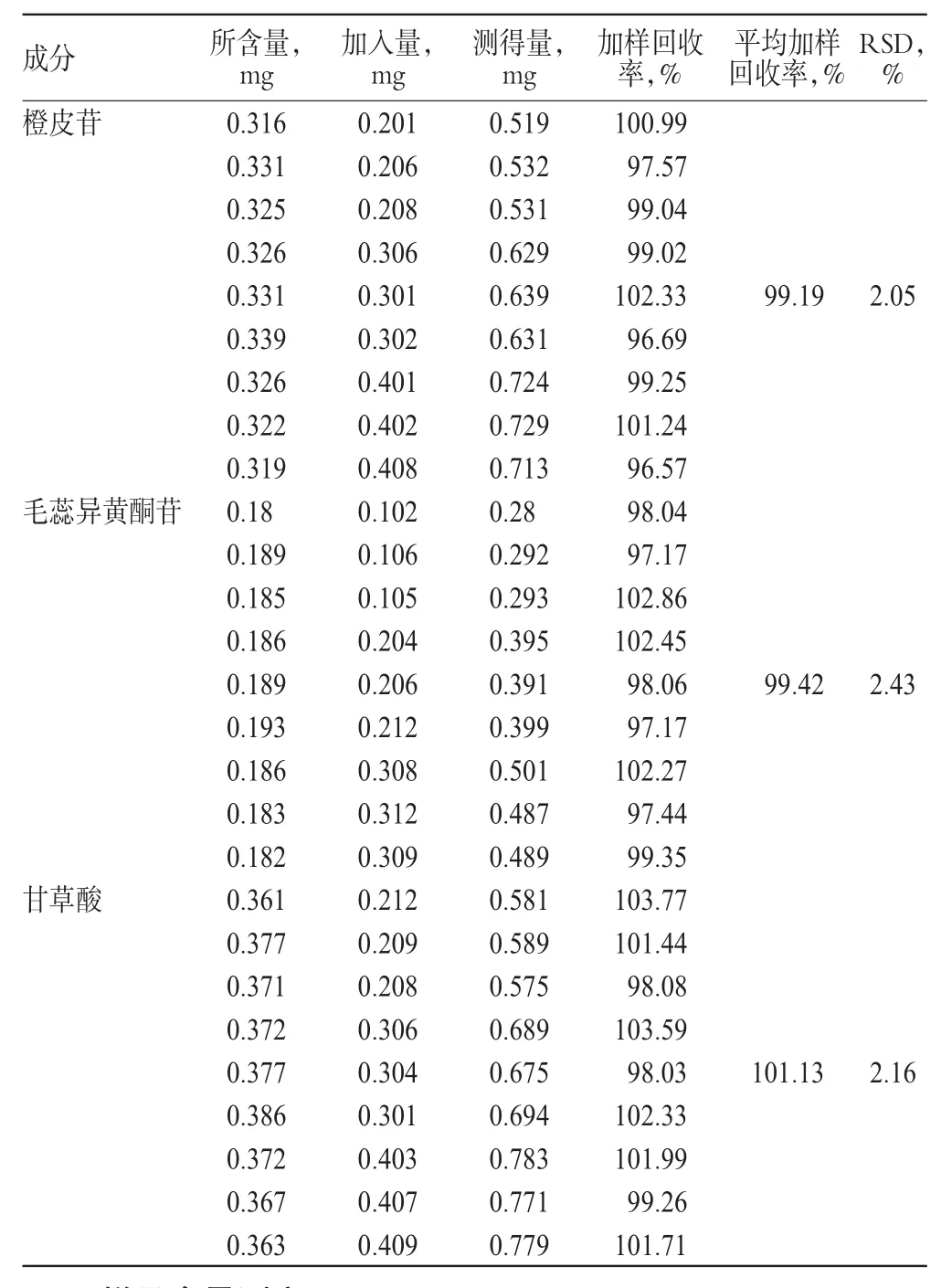
表3 加样回收率试验结果(n=9)Tab3 Results of recovery test(n=9)
2.9 样品含量测定
取7批补中益气丸样品各适量,分别按“2.2.1”项下方法制备供试品溶液,并按“2.1”项下色谱条件进样测定,计算3种成分含量,结果见表4。
3 讨论
3.1 流动相的选择
笔者曾尝试了分别采用甲醇-水与乙腈-水按不同的比例作为流动相进行测定,结果发现,3种待测成分的分离度均不理想,目标峰拖尾较严重;又尝试改为甲醇-0.1%磷酸溶液、乙腈-0.1%磷酸溶液作为流动相进行测定,结果发现,甲醇-0.1%磷酸溶液作为流动相进行梯度洗脱时,3种待测成分分离度均较好且峰形最佳。因此,选择甲醇-0.1%磷酸溶液作为流动相。
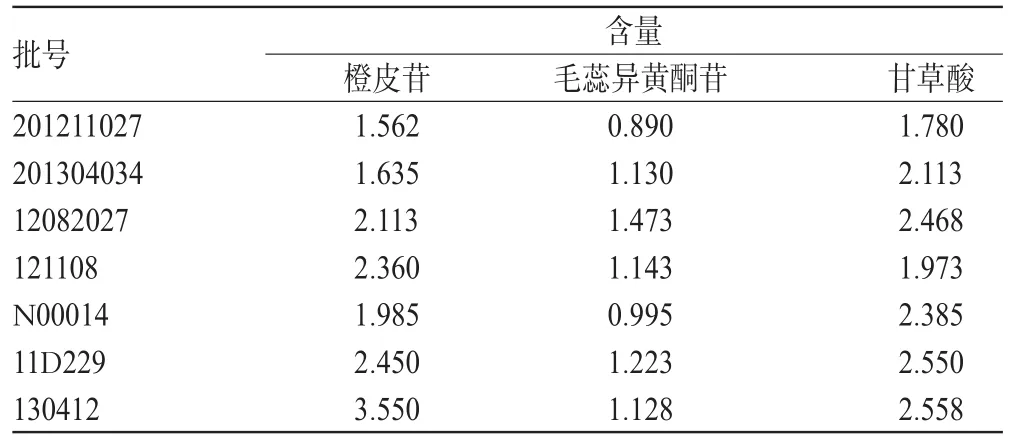
表4 样品含量测定结果(mg/g,n=3)Tab4 Results of content determination of samples(mg/g,n=3)
3.2 检测波长的选择
取对照品溶液在210~400nm波长范围内进行检测,结果可见,橙皮苷在280nm波长附近处有最大吸收,毛蕊异黄酮苷在260nm波长处有最大吸收,甘草酸在240nm波长处有最大吸收。当在260nm波长时,3种待测成分分离度均良好,可满足定量测定要求,故选择260nm为检测波长。
3.3 柱温的选择
取供试品溶液在选定的流动相及波长条件下观察了20、25、30、35℃等不同柱温对各成分分离效果的影响,结果发现,当柱温为30℃时,3种待测成分分离度均较好且峰形最佳。
综上所述,本方法准确、可靠、简便易行,可用于补中益气丸的质量控制。
[1] 刘晓玲,王汝俊,付铨盛.补中益气汤对脾虚大鼠胃黏膜MEK/ERK mRNA表达的影响[J].中药药理与临床,2013,29(1):5.
[2] 高璟春,张金超,陈瑶,等.补中益气汤的LC-MS分析及其对免疫抑制小鼠的调节作用[J].中草药,2006,37(8):1134.
[3] 许琦,王建华,王汝俊,等.补中益气汤对脾虚大鼠胃泌素受体结合作用的影响及其对胃黏膜损伤的保护机制[J].广州中医药大学学报,2003,20(1):51.
[4] 曾昭明,陈芝喜,赵慧,等.补中益气丸对脾虚大鼠甲状腺激素水平的影响[J].广州中医药大学学报,2007,24(4):320.
[5] 国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:780-781.
[6] 李洪刚,熊胜元,杨修齐,等.高效液相色谱法测定补中益气丸中甘草酸含量[J].中国药业,2005,14(7):43.
[7] 李苑,柯雪红,陈为,等.补中益气汤不同配伍对橙皮苷含量的影响[J].中药新药与临床药理,2009,20(3):262.
[8] 徐端琼.高效液相色谱法测定补中益气丸中橙皮苷的含量[J].海峡药学,2007,19(3):48.
[9] 柯雪红,花汝风,陈锦富,等.补中益气汤中甘草酸含量测定及配伍对其影响[J].中成药,2009,31(2):245.
Simultaneous Determination of 3Active Constituents in Buzhong Yiqi Pills by HPLC
XUE Wei1,MAO Ju-hua2,WANG Zhi-fang3
(1.Dept.of Pharmacy,Ruikang Hospital of Guangxi University of TCM,Nanning 530011,China;2.Lishui Institute for Food and Drug Control,Zhejiang Lishui 323000,China;3.Guangzhou Xiangxue Pharmaceutical Co.,Ltd.,Guangzhou 510663,China)
OBJECTIVE:To establish a method for simultaneous determination of hesperidin,calycosin glycoside and glycyrrhizic acid in Buzhong yiqi pills.METHODS:HPLC method was adopted.The determination was performed on Kromasil C18column with mobile phase consisted of methanol-0.1%phosphoric acid(gradient elution)at the flow rate of 1.0mL/min.The column temperature was 30℃,and the detection wavelength was set at 260nm.The sample size was 10μl.RESULTS:The linear ranges were 0.0615-1.250mg/ml for hesperidin(r=0.9997),0.0255-0.510mg/ml for calycosin glycoside(r=0.9998)and 0.0506-1.011mg/ml for glycyrrhizic acid(r=0.9999);RSDs of precision,stability and reproducibility tests were lower than 0.98%.Average recoveries were 99.19%(RSD=2.05%,n=9),99.42%(RSD=2.43%,n=9)and 101.13%(RSD=2.16%,n=9).CONCLUSIONS:The method is accurate,reliable,simple and feasible,and can be used for the quality control of Buzhong yiqi pills.
HPLC;Buzhong yiqi pills;Hesperidin;Calycosin glycoside;Glycyrrhizic
R917
A
1001-0408(2014)16-1524-03
DOI10.6039/j.issn.1001-0408.2014.16.30
*硕士。研究方向:医院药学。电话:0771-2188297。E-mail:xw5346@foxmail.com
2013-12-06
2014-01-05)
·药学教育·
