丙戊酸钠对肺癌细胞MICA蛋白表达的影响及其信号转导机制*
2014-01-01周向东陈贵华蒋幼凡
梅 花, 周向东, 陈贵华, 蒋幼凡
重庆医科大学附属第二医院呼吸内科,重庆 400010
肺癌等恶性肿瘤的发生、发展与体内的细胞免疫失调有着密切联系。研究表明,在肺癌等恶性肿瘤的发生发展中能检测到人类MHCⅠ类分子链相关基因 A(human MHC classⅠchain-related A,MICA)蛋白的高表达[1-2],而体内的γδT 细胞、自然杀伤(natural killer,NK)细胞等免疫活性细胞,可通过表面受体——NK细胞表面受体(NKG2D)与MICA特异性结合,启动细胞毒性作用,导致肺癌等肿瘤细胞死亡[2-3]。大量研究发现肿瘤细胞通过脱落表面膜MICA,逃避γδT细胞、NK细胞的细胞毒性作用[4-5]。如何增加肺癌等肿瘤细胞 MICA蛋白的表达及减少MICA蛋白的脱落进而增加γδT细胞、NK细胞等的细胞毒性作用,是现阶段肿瘤细胞免疫研究的重点。近期研究表明组蛋白去乙酰酶抑制剂能通过增加细胞内组蛋白的乙酰化程度,上调肺癌等肿瘤细胞MICA蛋白的表达[6],但是组蛋白去乙酰酶抑制剂如何增加MICA蛋白的表达,其增加MICA蛋白表达的具体作用机制少有涉及。诸多基于上皮以及内皮细胞增殖的研究表明,MICA的表达升高依赖于 MAPK家族(ERK1/2、JNK、p38MAPK)的 激 活[7-8]。 本 研 究 使 用 丙 戊 酸 钠(VAP)作为组蛋白去乙酰酶抑制剂,研究其对肺癌A549细胞MICA蛋白的影响,并通过探讨MAPK主要信号通路在VPA刺激A549细胞MICA蛋白表达影响中的作用,为从分子信号链接机制角度研究组蛋白去乙酰酶抑制剂增强MICA蛋白的表达提供理论及实验依据,并为组蛋白去乙酰酶抑制剂在γδT细胞、NK细胞基于NKG2DMICA途径介导的肺癌等肿瘤细胞免疫中的运用提供理论依据。
1 材料与方法
1.1 材料
肺癌A549细胞购于美国ATCC公司,RPMI 1640干粉及胎牛血清、双抗(青霉素、链霉素)购自HyClone公司,VPA、ERK1/2抑制剂(PD98059)、p38蛋白 激 酶 抑 制 剂 (SB203580)、JNK 抑 制 剂(SP600125)购自于Sigma公司,兔抗人MICA一抗购于美国Abcom公司,山羊抗兔二抗及内参兔抗人β-actin一抗购自北京四正柏生物科技有限公司,IP蛋白裂解液、BCA蛋白浓度测定试剂盒、SDSPAGE配胶试剂盒、ECL发光试剂盒购自南京凯基生物科技发展有限公司,MICA基因PCR引物由大连宝生物公司合成,Trizol RNA提取试剂盒、Taq DNA聚合酶、dNTP、DNA Marker DL1000、琼脂糖均购自TaKaRa公司。
1.2 实验方法
1.2.1 肺癌A549细胞的培养 A549细胞株培养于50mL玻璃培养瓶中,加入3mL含10%胎牛血清的RPMI 1640培养液(双抗浓度为100U/mL),置于5%CO2、37℃恒温培养箱中培养,细胞每2天换液1次,显微镜下观察细胞密度为80%左右时传代培养,培养细胞生长状态至对数生长期,使用RPMI 1640调整细胞密度至106/mL备用。
1.2.2 不同浓度的VPA处理肺癌A549细胞 将对数生长期的肺癌A549细胞以5×105/孔种植于6孔板中,每孔中加入2mL培养液,并在6孔板中分别加入不同浓度的VPA,使VPA终浓度达到0.1、0.5、1.0、2.0mmol/L,并设置加入等体积纯溶剂的空白对照组。在5%CO2、37℃恒温培养箱中培养24h。使用RT-PCR技术检测A549细胞 MICA mRNA转录水平,使用 Western blot法检测A549细胞MICA蛋白表达水平。每组实验设置5个复孔,独立实验重复5次用于统计学分析。利用Quantity one软件分析灰度值,检测出最佳VPA刺激浓度。
1.2.3 实验分组 将处于对数生长期的肺癌A549细胞以5×105/孔种植于6孔板中。相同培养条件下,将A549细胞随机分为5组:①空白对照组;②VPA组;③VPA+ PD98059组;④VPA+SB203580组;⑤VPA+SP600125组。各组设置5个复孔,VPA浓度为1.0mmol/L(依据1.2.2部分实验得出VPA浓度在1.0mmol/L时能最大化刺激 MICA 蛋 白 表 达 ),PD98059、SB203580、SP600125各抑制剂终浓度均为20μmol/L。RPMI 1640调整每孔培养液体积均为2mL。5%CO2、37℃恒温培养箱中培养24h。使用RT-PCR法及Western blot法分别检测A549细胞MICA mRNA以及蛋白水平。ELISA法检测细胞培养液中分泌型MICA(sMICA)的含量,独立实验重复5次用于统计学分析。
1.2.4 RT-PCR技术检测 A549细胞 MICA mRNA转录水平 将上述处理的A549细胞用Trizol法提取总RNA,紫外分光光度仪测得A260nm/A280nm值波动于1.80~2.20。再参照TaKaRa逆转录试剂盒步骤制取cDNA,两步法进行扩增。引物序列如下:目的基因 MICA 引物 (296bp),5′-TCTCCCAAAACCTGGAGACTAA-3′(上 游 ),5′-CCAGCTCAGTGTGATATTCCAG-3′(下 游);内 参β-actin 引 物 (564bp),5′-CTGGGACGACATGGAGAAA-3′(上 游 ),5′-AAGGAAGGCTGGAAGAGTGC-3′(下游)。参照说明书配制扩增体系,扩增条件如下:94℃预变性5min、94℃变性30s、58℃退火(内参59℃退火)30s、72℃延伸30s(内参延伸50s)、35个循环(内参28个循环),72℃延伸5 min。扩增产物进行3%的琼脂糖凝胶电泳,BioRad成像仪成像,并拍照分析。以“MICA mRNA吸光度值/内参吸光度值”作半定量分析。
1.2.5 Western blot检测A549细胞 MICA蛋白的表达水平 参照说明书取上述处理的A549细胞,提取总蛋白(在冰上操作),BCA法检测总蛋白浓度。加入上样Buffer,调整总蛋白浓度至4μg/μL,沸水中加热5min。参照说明书配制10%的分离胶及5%浓缩胶,每孔加入15μL蛋白溶液(60 μg/孔),SDS-PAGE凝胶电泳,切胶250mA 恒流电转至PVDF膜。采用PBST配制的5%脱脂奶粉封闭1h,PBST洗膜1次。以5%脱脂奶粉按照1:1 000稀释 MICA一抗(β-actin内参1:3 000),4℃冰箱过夜孵育。次日PBST洗膜3次,每次10 min,用5%脱脂奶粉按照1:5 000稀释HRP标记山羊抗兔二抗,37℃孵育1h。PBST洗膜3次,每次10min。ECL发光法成像并拍照分析。
1.2.6 ELISA法检测A549细胞培养上清液中分泌型MICA(sMICA)蛋白水平 取细胞培养上清液,以3 000r/min离心15min(离心半径13.5 cm),取上清液,按人 MICA ELISA检测试剂盒说明书进行上清液中的sMICA测定。
1.3 统计学分析
各检测组 Western blot条带及RT-PCR条带均采用Quantity one软件分析,实验数据使用SPSS 18.0软件进行统计学分析,样本均数的两两比较采用t检验,多组间均数比较采用方差分析,结果以±s表示,以P<0.05为差异具有统计学意义。
2 结果
2.1 VPA对A549细胞MICA mRNA水平的影响
加入不同浓度的VPA刺激24h后,使用RTPCR技术检测各组细胞MICA mRNA水平差异。结果显示,各组MICA mRNA转录水平较空白对照组均有显著增加(均P<0.05);除 VPA浓度2.0 mmol/L 组 [(0.96±0.06)]与 1.0mmol/L 组[(0.94±0.08)]MICA mRNA 水平差异无统计学意义(P>0.05)外,各浓度组间 MICA mRNA水平两两比较差异均有统计学意义(均P<0.05),呈现浓度依赖性。见图1。
2.2 VPA对A549细胞MICA蛋白合成的影响
加入不同浓度的VPA刺激24h后,使用Western blot法检测各组A549细胞MICA蛋白的表达水平。结果显示,各组MICA蛋白的表达均较空白对照组明显增加(均P<0.05);除VPA浓度为1.0mmo/L、2.0mmol/L组间 MICA 蛋白量差异无统计学意义(P>0.05)外,各浓度组间 MICA蛋白水平两两比较差异均有统计学意义(均P<0.05),呈现浓度依赖性。见图2。

图1 不同浓度VPA对MICA转录的影响Fig.1 The transcription levels of MICA mRNA induced by different concentrations of VPA

图2 不同浓度VPA对MICA合成的影响Fig.2 The synthesized levels of MICA induced by different concentrations of VPA
2.3 MAPK信号通路抑制剂对A549细胞MICA mRNA及蛋白水平的影响
各实验组分别加入MAPK信号通路相关抑制剂预处理后,给予1.0mmol/L VPA 培养24h,使用RT-PCR技术检测A549细胞MICA mRNA水平变化,使用 Western blot技术检测 A549细胞MICA蛋白合成量。结果显示,分别加入ERK抑制剂PD98059、p38蛋白激酶抑制剂SB203580后,VPA(1.0mmo/L)对肺癌 A549细胞 MICA 在基因的转录水平及表达水平的增加较未加入抑制剂组明显减弱(P<0.05),而加入JNK抑制剂SP600125后,MICA蛋白的表达无明显改变(P>0.05),见图3。

图3 MAPK信号通路抑制剂预处理对各组VPA刺激后MICA mRNA以及蛋白表达的影响Fig.3 The influences of pretreatment of inhibitors of MAPK signal pathway on expression of MICA induced by VPA
2.4 ELISA法检测细胞培养上清液中sMICA水平
A549细胞上清液中的sMICA水平,采用ELISA法检测。结果表明,使用0.1~2.0mmol/L VPA刺激细胞24h并不影响培养上清液中的sMICA浓度(P>0.05)。使用MAPK相关信号通路抑制剂预处理后,再给予VPA刺激24h,各组A549细胞培养上清液中sMICA浓度差异亦无统计学意义(P>0.05)。见图4。
3 讨论
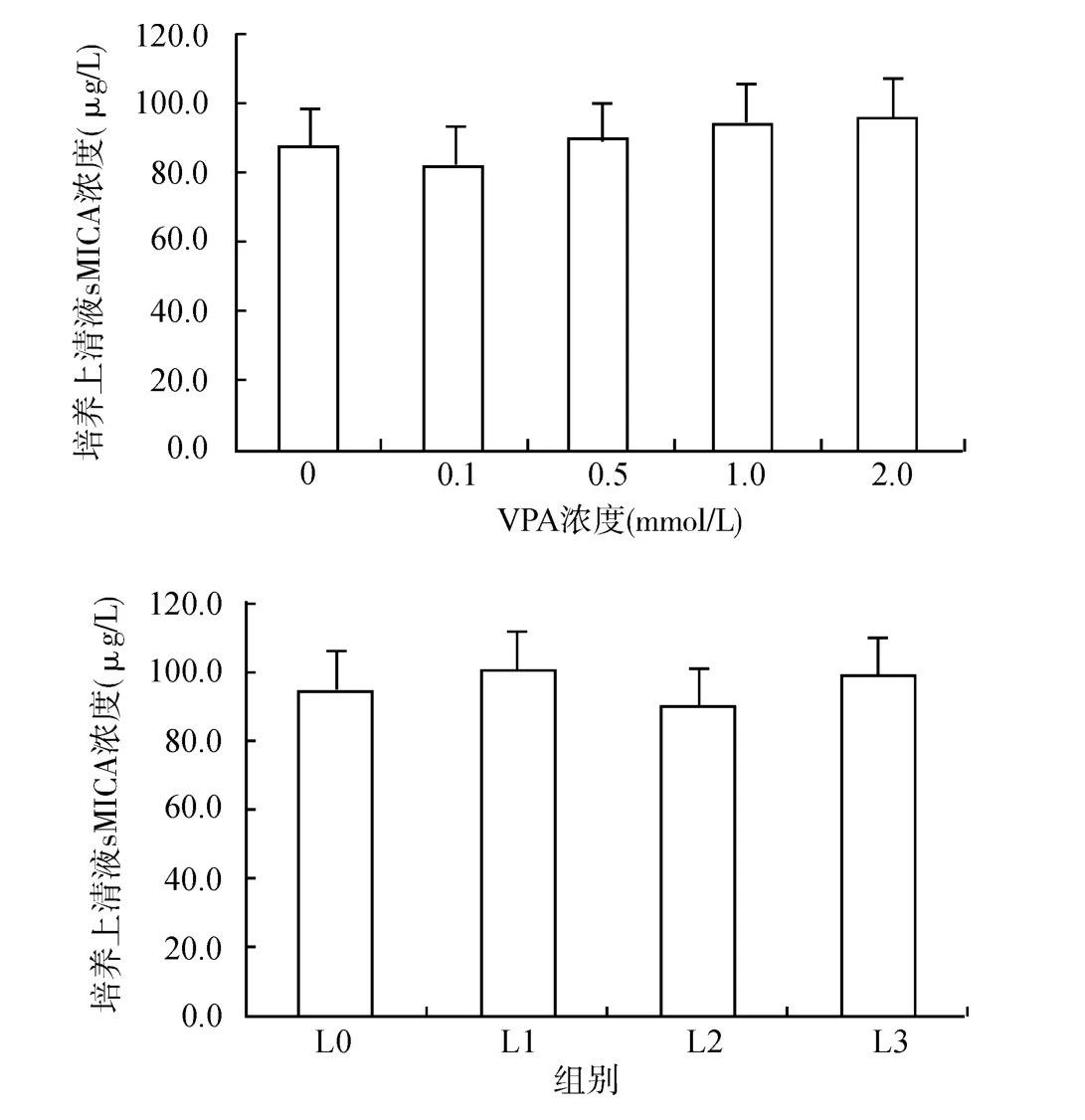
图4 ELISA法检测A549细胞上清液中sMICA浓度Fig.4 Concentration of sMICA in cell culture supernatant of A549cells
肺癌是目前导致死亡的主要恶性肿瘤之一。在传统的化疗、放疗、手术等治疗手段外,肿瘤的细胞免疫也日趋受到关注。体内外研究显示γδT细胞、NK细胞可通过表面受体NKG2D与肿瘤细胞表面的MICA配体特异性结合,诱发肿瘤细胞发生凋亡[1-3]。研究显示 γδT 细胞属于 CD4、CD8双阴性细胞,其介导的肿瘤免疫不受MHC限制,对肿瘤的免疫介于适应性免疫与固有免疫之间[9],大量的研究显示肺癌等肿瘤细胞可通过脱落MICA蛋白逃避或者降低γδT细胞、NK细胞的细胞毒性作用[4-5],并且肿瘤恶性程度及侵袭力的高低与 MICA脱落程度密切相关。
本实验结果显示,VPA能正向调节肺癌A549细胞内MICA基因的转录及蛋白合成,并在一定范围内呈浓度依赖性,这与一些国内外肿瘤细胞学的相关报道一致[6,10]。然而对于VPA正向调节肺癌细胞MICA表达的机制目前尚不清楚,鉴于已有的基于上皮细胞以及内皮细胞中MICA调控机制的相关报道,本研究从MAPK信号通路的3条主要途径,即 ERK1/2、JNK、p38[7-8,11]着手,探讨 VPA 的调控机制。通过相关信号节点分子的特异性抑制剂预处理,我们发现VPA主要通过ERK及p38 MAPK信号通路影响肺癌A549细胞MICA蛋白的表达,而非JNK依赖性信号通路;并且阻断ERK、p38MAPK任意一条通路,均能显著抑制VPA诱导的MICA蛋白的高合成。这与一些关于VPA在神经系统中分子药理机制的研究相符[12-13]。ERK1/2、p38MAPK信号通路影响肿瘤细胞 MI-CA的合成水平也在其它器官来源的肿瘤细胞研究中有所报道[8]。近年来,VPA抗肿瘤的作用也为诸多研究所关注,除了作为组蛋白去乙酰化酶抑制剂直接抑制肿瘤细胞生长外,多条信号通路及基因、细胞因子的不同作用方式也不断被揭示。大量的研究显示 VPA 提高了 肺癌、肝癌[3,8,10]等肿 瘤 细 胞 的MICA蛋白的表达,从而提高了γδT细胞、NK细胞的细胞毒性作用,一些临床研究也显示VPA联合治疗能延长肿瘤晚期患者的生存时间,对肿瘤的治疗起到了一定的辅助作用。
分泌型MICA(sMICA)为细胞表面膜结合型(mMICA)的脱落产物。血浆中的sMICA不仅降低了肿瘤细胞表面的mMICA水平,减低了肿瘤细胞的免疫原性。同时,过量的sMICA能全面下调NK细胞、T细胞、巨噬细胞表面的NKG2D受体,并下调其细胞免疫活性[14]。在一定程度上,膜型/分泌型MICA比值,影响着NK细胞、γδT细胞、巨噬细胞等肿瘤免疫细胞的识别功能,在一定程度上决定了其肿瘤免疫学功能[15]。本研究证实,VPA刺激可提高细胞中MICA表达,但不影响肺癌细胞A549培养上清液中sMICA的水平,不会导致sMICA的增高带来的肿瘤“免疫逃避”的增加。因此,从体外细胞实验的角度分析,VPA有希望成为理想的肿瘤细胞免疫的“增敏剂”,并可能为今后肿瘤的细胞免疫治疗提供新的思路。
[1] 王义平,张彩,牛家峰,等.NKG2D配体在13种肿瘤细胞系中的表达及意义[J].癌症,2008,27(3):243-248.
[2] Ashiru O,Boutet P,Fernández-Messina L,et al.Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2Dligand MICA*008that is shed by tumor cells in exosomes[J].Cancer Res,2010,70(2):481-489.
[3] Bennouna J,Bompas E,Neidhardt E M,et al.Phase-Ⅰ study of Innacell gammadelta,an autologous cell-therapy product highly enriched in gamma9delta2Tlymphocytes,in combination with IL-2,in patients with metastatic renal cell carcinoma[J].Cancer Immunol Immunother,2008,57(11):1599-1609.
[4] Marincola F M,Jaffee E M,Hicklin D J,et al.Escape of human solid tumors from T-cell recognition:molecular mechanisms and functional significance[J].Immunol,2000,74(6):181-273.
[5] Wu J D,Higgins L M,Steinle A,et al.Prevalent expression of the immunostimulatory MHC classⅠ chain-related molecule is counteracted by shedding in prostate cancer[J].J Clin Invest,2004,114(4):560-568.
[6] Armeanu S,Bitzer M,Lauer U M,et al.Natural killer cellmediated lysis of hepatoma cells via specific induction of NKG2Dligands by the histone deacetylase inhibitor sodium valproate[J].Cancer Res,2005,65(14):6321-6329.
[7] Lim J H,Woo J S,Shin Y W.Cilostazol protects endothelial cells against lipopolysaccharide-induced apoptosis through ERK1/2-and P38MAPK-dependent pathways[J].Korean J Intern Med,2009,24(2):113-122.
[8] Wu X,Tao Y,Hou J,et al.Valproic acid upregulates NKG2D ligand expression through an ERK-dependent mechanism and potentially enhances NK cell-mediated lysis of myeloma[J].Neoplasia,2012,14(12):1178-1189.
[9] Holtmeier W,Kabelitz D.Gammadelta T cells link innate and adaptive immune responses[J].Chem Immunol Allergy,2005,86(11):151-183.
[10] Glaser K B,Staver M J,Waring J F,et al.Gene expression profiling of multiple histone deacetylase(HDAC)inhibitors:defining a common gene set produced by HDAC inhibition in T24and MDA carcinoma cell lines[J].Mol Cancer Ther,2003,2(2):151-163.
[11] 刘萍,邓元俊,裴广畅,等.ERK1/2信号通路的活化参与人脐静脉内皮细胞向间充质转分化的过程[J].华中科技大学学报:医学版,2013,42(5):505-510.
[12] 朱锦莉.P38MAPK在PTZ致痫大鼠中的激活及丙戊酸钠的干预作用[J].辽宁医学院学报,2012,33(1):19-22.
[13] 李建玲,汤参娥,陈主初,等.丙戊酸钠活化大鼠海马和额叶ERK-1/2信号传导通路[J].生物化学与生物物理进展,2003,30(2):239-244.
[14] Groh V,Wu J,Yee C,et al.Tumour-derived soluble MIC ligands impair expression of NKG2Dand T-cell activation[J].Nature,2002,419(6908):734-738.
[15] Salih H R,Rammensee H G,Steinle A.Cutting edge:downregulation of MICA on human tumors by proteolytic shedding[J].J Immunol,2002,169(8):4098-1102.
