比阿培南开环物杂质的确证及含量测定Δ
2013-12-03陈松杰王德刚钟雅妮珠海联邦制药股份有限公司广东中山528467
陈松杰,王德刚,张 莉,钟雅妮(珠海联邦制药股份有限公司,广东中山 528467)
比阿培南(Biapenem)是一种新型1β-甲基碳青霉烯类抗生素,具有抗菌谱广、对肾脱氢肽酶稳定、药动学参数优良、临床治疗效果好、耐受性好和不良反应少等特点[1]。目前各国药典均未收载比阿培南。经分析其主要降解产物为聚合物和开环物,文献[2-6]中多对其聚合物进行研究,对其开环物的研究鲜有报道。而从比阿培南聚合物的产生途径可知,其开环物可与比阿培南反应生成二聚物,故对开环物进行控制可在一定程度上降低聚合物产生的可能性。
笔者另查阅注射用Omegacin的说明书[7]知,原研品中可能存在的杂质包括2个开环物,现暂命名为比阿培南开环物A和开环物B,且开环物A为比阿培南体内代谢产物。本文通过试验建立了测定比阿培南开环物杂质的反相高效液相色谱(RP-HPLC)法,并采用色谱-质谱(LC-MS)方法对其结构进行确证,同时考察了自制样品及原研品中开环物杂质的含量水平。
1 材料
LC-20AT型HPLC仪、SPD-M20A二极管阵列检测器(日本Shimadzu公司);AE240型电子天平(瑞士Mettler toledo公司);6120型单四级杆液质联用仪(美国Agilent公司)。
比阿培南原料药(自制,批号:4031204001、4031204002、4031204003,纯度:99.96%、99.96%、99.95%);注射用比阿培南(原研品,日本明治制果制药株式会社,批号:4004,测定时已近效期,规格:每支0.3 g);比阿培南开环物B杂质对照品(自制,经质谱、元素分析及核磁共振氢谱确证其结构,批号:120703,纯度:99.8%);由于比阿培南开环物A杂质对照品获得较为困难,因此拟取其HPLC洗脱成分采用LC-MS方法确证其结构;乙腈为色谱纯,水为自制超纯水,其他各试剂均为分析纯。
2 方法与结果
2.1 色谱条件
HPLC色谱条件:色谱柱为Kromasil 100-5 C18(250 mm×4.6 mm,5 μm),流动相为0.01 mol/L四丁基溴化铵溶液-乙腈-三乙胺(980∶20∶2,用磷酸调节pH值至6.0),流速为1.0 ml/min,检测波长为254 nm,柱温为30℃。
LC-MS色谱条件:色谱柱为GL Sciences C18(250 mm×4.6 mm,5 μm),流动相A为0.01 mol/L醋酸铵溶液,流动相B为乙腈,梯度洗脱,流速为1.0 ml/min,柱温为30℃。按表1进行线性梯度洗脱。

表1 线性梯度洗脱表Tab 1 Linear gradient elution
质谱条件:电喷雾离子源(ESI);正离子模式采集数据;选择一定质荷比(m/z)范围内的全扫描,m/z为100~800;氮气流速为10 L/min,雾化器压力为35 psig,干燥气体温度为300℃,毛细管电压为4.0 kV。
2.2 溶液的制备
2.2.1 系统适用性试验溶液制备。取比阿培南原料药(批号:4031204001)约15 mg,置于10 ml量瓶中,加适量流动相溶解后,于80℃水浴破坏15 min,放冷至室温,加5%甲酸溶液0.5 ml,再加流动相稀释至刻度,摇匀,即得含有比阿培南2个开环物杂质的溶液,作为系统适用性试验溶液。
2.2.2 供试品溶液的制备。取供试样品适量,精密称定,加流动相溶解并定量稀释制成每1 ml中含1.5 mg的溶液,作为供试品溶液(临用新制)。
2.2.3 对照溶液的制备。精密量取供试品溶液适量,加流动相定量稀释制成15 μg/ml的溶液,即得。
2.2.4 比阿培南开环物A定位溶液的制备。取系统适用性试验溶液继续水浴破坏,同时增加进样体积,在检测器出口处收集第1个峰面积显著增大的杂质的洗脱成分,即得。
2.2.5 比阿培南开环物B对照品溶液的制备。取比阿培南开环物B杂质对照品适量,加流动相溶解,摇匀,即得。
2.3 系统适用性试验溶液破坏条件的确定
比阿培南结构中的内酰胺环不稳定,在高温、酸或碱条件下易开环,降解为比阿培南开环物A;且开环后产生的—COOH基不稳定易脱去生成甲酸,甲酸使比阿培南开环,并与五元环上的—NH结合产生开环物B。因此选择一定量的甲酸为破坏溶剂进行试验。取系统适用性试验溶液20 μl注入色谱仪,记录色谱图,主峰后峰面积显著增大的2个杂质峰依次为比阿培南开环物A、B。主峰与比阿培南开环物A色谱峰的分离度应大于1.5。
比阿培南可能降解的途径如图1所示:

图1 比阿培南开环物可能的降解途径Fig 1 Potential degradation pathway of biapenem openring impurities
2.4 比阿培南开环物的结构确证
取系统适用性试验溶液,进行LC-MS联用分析,结果检出2个较大的降解杂质峰,保留时间约为3.8 min的杂质准分子离子峰的m/z分别为369、325,与[M+H]+、[M-COOH+H]+对应,即m/z为369的峰分子质量为368,与比阿培南开环物A的分子质量368一致;m/z为325的峰为比阿培南开环物A的碎片离子,因比阿培南开环后—COOH基易脱去,形成该碎片离子;保留时间约为5.4 min处的杂质准分子离子峰[M+H]+的m/z为397,即m/z为397的峰分子质量为396,与比阿培南开环物B分子质量396一致,且其保留时间及m/z均与比阿培南开环物B对照品一致。表明系统适用性试验溶液中降解产生的2个较大杂质分别为比阿培南开环物A、B,详见图2。
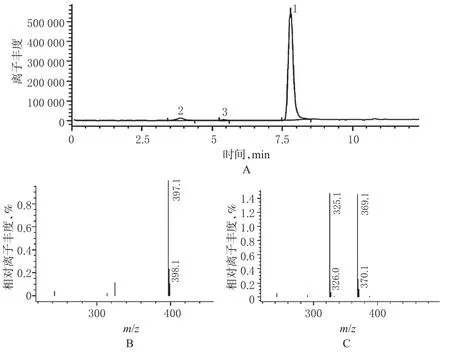
图2 降解杂质经LC-MS结构确证分析结果A.总离子流图;B.开环物B的MS图;C.开环物A的MS图;1.比阿培南;2.开环物A;3.开环物BFig 2 LC-MS analysis results of structural identification of impurity by degradationA.TIC;B.MS chromatograms of open-ring impurity B;C.MS chromatograms of open-ring impurity A;1.biapenem;2.open-ring impurity A;3.open-ring impurity B
2.5 比阿培南开环物在2种色谱系统中的相互识别
通过对比2种色谱系统中杂质含量,结果系统适用性试验溶液在HPLC色谱系统中的2个峰面积显著增大的杂质,分别与LC-MS系统下峰面积显著增大的杂质含量基本吻合,两者存在对应关系;另采用开环物B杂质对照品在HPLC色谱系统进行定位,可证明第2个显著增大的杂质即为比阿培南开环物B;收集HPLC色谱系统系统适用性试验溶液中第1个显著增大杂质的洗脱成分,再在LC-MS系统中进样测试,其保留时间与比阿培南开环物A一致,且其分子质量为368,与比阿培南开环物A分子质量亦一致,可证明第1个显著增大的杂质即为比阿培南开环物A。
由此说明,LC-MS系统检出分子质量为368和396的杂质,分别与HPLC色谱系统检出的2个杂质为同一物质,即HPLC色谱系统中此2个杂质分别为比阿培南开环物A、B。
2.6 比阿培南开环物A、B的含量测定
2.6.1 系统耐用性试验及开环物峰位的确定。照“2.1”项下HPLC色谱条件,取系统适用性试验溶液,分别采用Thermo Hypersil ODS C18(250 mm×4.6 mm,5 μm)、Kromasil 100-5 C18(250 mm×4.6 mm,5 μm)和大连依利特ODS C18(250 mm×4.6 mm,5 μm)3种不同厂家色谱柱,在不同仪器上进行试验。结果系统适用性试验溶液中开环物A与比阿培南的分离度均大于1.5,开环物B保留时间与开环物B对照品一致;且比阿培南开环物A和B均增加明显,便于对杂质进行定位,方法可行,耐用性好。因此采用系统适用性试验溶液对开环物进行定位。
2.6.2 专属性试验及开环物降解途径考察。取比阿培南原料药(批号:4031204001)适量,分别经水浴和5%甲酸溶液(系统适用性试验溶液)、高温(80℃)、光照[(4500±500)lx]、高湿(相对湿度92.5%)及原料药溶液经85℃水浴破坏后制成的溶液进样分析;同时取比阿培南开环物B对照品适量,加流动相制成一定质量浓度的溶液进样分析,记录色谱图,考察方法专属性。空白(流动相)、开环物B对照品及样品经高温、高湿、光照、水浴及强酸破坏后的色谱见图3。

图3 方法专属性考察HPLC图谱A.空白(流动相);B.样品;C.系统适用性试验溶液;D.高温破坏后样品;E.光照破坏后样品;F.高湿破坏后样品;G.水浴破坏后样品;H.开环物B对照品;1.比阿培南;2.开环物A;3.开环物BFig 3 HPLC chromatograms of method specificityA.blank(mobile phase);B.sample;C.solution of system suitability test;D.sample destroyed by high temperature;E.sample destroyed by light;F.sample destroyed by humidity;G.sample destroyed by water bath;H.open-ring impurity B control;1.biapenem;2.open-ring impurityA;3.open-ring impurity B
结果表明,样品水溶液在水浴破坏条件下,开环物A增加显著,但未降解产生开环物B;经水浴及强酸破坏(即系统适用性试验溶液)后,开环物A及开环物B均增加显著;高温、高湿及光照条件下开环物A、B均未显著增加,表明在此几种条件下开环物不易产生。降解产生的开环物B杂质与其对照品保留时间一致;空白溶剂不影响比阿培南及其开环物的检出;各降解杂质均能与开环物分离完全。本色谱系统专属性良好。因系统适用性试验即是在酸性环境下使比阿培南水解,试验结果表明水解产物对测定无干扰,故专属性试验不再考察酸碱水解对测定的干扰。
2.6.3 线性关系试验。精密称取比阿培南原料药(批号:4031204001)和比阿培南开环物B对照品适量,加流动相溶解后,再加流动相定量稀释制成系列质量浓度分别为0.077、0.155、0.309、0.464、0.773、1.237、1.546 mg/ml和 0.765、1.53、3.06、4.59、7.65、12.24、15.30、22.95 μg/ml的混合溶液,摇匀,即为线性试验溶液。进样测试,将测得的峰面积(y)与质量浓度(x)作线性回归。结果比阿培南的线性方程为:y=758510x-1021662(r=0.9955);比阿培南开环物B的线性方程为:y=1172380x-2703(r=0.9999)。表明比阿培南与比阿培南开环物B检测质量浓度线性范围分别为0.077~1.546 mg/ml、0.765~22.95 μg/ml。
2.6.4 定量限及检测限试验。取“2.6.3”项下线性试验溶液,逐步稀释制成一定质量浓度的溶液,进样测试。得比阿培南与比阿培南开环物B的定量限(信噪比约为10∶1)分别为2.1、2.3 ng,检测限(信噪比约为3∶1)分别为0.6、0.9 ng。
2.6.5 比阿培南开环物B回收率试验。精密称取比阿培南开环物B适量,加流动相溶解并制成0.15 mg/ml的溶液,作为杂质贮备液。再精密称取本品9份,各约30 mg,分别置于9个20 ml量瓶中,分3组,各3份,每组分别加入精密量取的杂质贮备液(0.15 mg/ml)1.6、2.0、2.4 ml,再加流动相溶解并稀释至刻度,摇匀,作为比阿培南开环物B回收率试验的供试品溶液(按比阿培南开环物B的限度为1.0%进行加样回收试验);另取杂质贮备液适量,加流动相制成0.015 mg/ml的溶液,作为比阿培南开环物B对照品溶液。精密量取上述供试品溶液及对照品溶液各20µl,分别注入色谱仪,记录色谱图。结果,比阿培南开环物B的平均回收率为100.5%,RSD=0.7%(n=3),样品对此杂质的测定无干扰,本方法准确性良好。
2.6.6 样品测定结果。取自制的3批比阿培南原料药及原研品1批,照“2.1”项下色谱条件,取对照溶液20 μl,注入色谱仪,调节检测灵敏度,使主峰的峰高约为满量程的20%;再精密量取供试品溶液和对照溶液各20 μl,分别注入色谱仪,记录色谱图至主峰保留时间的5倍。开环物A、B含量均采用自身对照法计算,结果见表2。

表2 4批样品开环物杂质的含量测定结果Tab 2 Content determination of open-ring impurities in 4 batches of samples
3 讨论
Nan Wang等[8]建立了比阿培南及杂质质控的HPLC分析方法,该方法利用HPLC-二极管阵列检测器(DAD)数据,采用二维色谱光谱相关分析技术及软件,在无杂质对照品的情况下,实现了质控分析HPLC-紫外(UV)色谱中的色谱峰与LCMS色谱图中的色谱峰的相互识别。结果表明比阿培南水解产生的杂质主要为开环物及二聚物,暂命名为比阿培南开环物A及C、比阿培南二聚物A及B。
本文则采用系统适用性试验溶液,在HPLC系统下即可对比阿培南开环物A及B进行定位,不依赖于LC-MS,更适用于日常检验。本文方法与文献[8]方法的条件比较见表3。
综上,本文建立的方法可对比阿培南的2个开环物杂质进行有效分离,方法专属强、灵敏度高,可满足杂质测定要求,因此本法可作为比阿培南中开环物的质控方法。同时,通过系统适用性试验,对特定杂质开环物A和B进行定位,不需要提供杂质对照品,操作方法简便,并可有效保证产品质量安全。

表3 2种分析方法的比较Tab 3 Comparison of 2 kinds of analysis methods
[1]赵敏,赵国君,张勇.新碳青霉烯类抗生素比阿培南[J].中国临床药理学杂志,2005,21(5):390.
[2]张菁,邢亮彬.HPLC法测定注射用比阿培南的含量及有关物质[J].中国抗生素杂志,2006,31(9):565.
[3]傅小雅.反相高效液相色谱法测定注射用比阿培南的含量及有关物质[J].海南医学院学报,2005,11(5):388.
[4]马杰,赵玉新,王广慧,等.凝胶色谱法测定比阿培南聚合物的含量[J].中国当代医药,2010,17(8):116.
[5]王蓓,张菁,马卫红.注射用比阿培南的有关物质测定方法研究[J].现代中西医结合杂志,2010,19(10):1247.
[6]刘俊华,张莉,程玉宝,等.HPLC法测定比阿培南二聚物方法研究及其降解途径考察[J].药物分析杂志,2012,32(3):468.
[7]明治制果制药株式会社.注射用Omegacin 0.3g[EB/OL].(2013-02)[2013-04-15].http://www.info.pmda.go.jp/.
[8]Nan Wang,Chun-Feng Li,Dou-Sheng Zhang,et al.Establishment of an HPLC method for the analysis of biapenem an its impurities[J].Journal of Chinese Pharmaceutical Sciences,2011,20(2):171.
