超高效液相色谱-串联质谱测定污泥中氯霉素、磺胺类、喹诺酮类、四环素类与大环内酯类抗生素
2013-11-28王硕,张晶,邵兵*
王 硕,张 晶,邵 兵*
(1.北京市朝阳区疾病预防控制中心,北京 100021;2.北京市疾病预防控制中心,食物中毒诊断溯源技术北京市重点实验室,北京 100013)
近年来,药品和个人护理品(PPCPs)作为环境新型污染物受到了广泛关注。药品中的抗生素主要用于预防和治疗细菌感染性疾病。据报道,仅2005年,我国生产约21万吨抗生素,其中9万吨作为兽药使用[1]。这些药物使用后,经动物排泄、废水处理等不同途径进入环境,从而可能引发细菌耐药性等一系列问题。目前在饮用水[2-3]、生活污水[4-5]、土壤、河流底泥和污泥[6-9]等多种环境中均检出了抗生素类药物残留。由于市政污水处理厂被认为是多种污染物消除的主要场所,且部分药物亲脂性较强,易在污泥体系中富集,因此,污泥中抗生素类药物的检出浓度可高达mg/kg级[7,10-11],给污泥堆肥、资源化使用和无害化处置带来了挑战。为了解污泥中多种抗生素的存在水平和污染现状,有必要建立一种同时测定污泥中多种抗生素药物的分析方法,以进行实际样品的监测。
本文选取50种常用的抗生素为研究对象,包括6种四环素、16种喹诺酮、20种磺胺类、7种大环内酯类药物以及氯霉素。由于污泥基质复杂,目标药物种类多且理化性质差异大,为了减少物质干扰、提高提取效率,样品的提取和净化步骤非常关键。对于污泥中药物等痕量污染物的提取,常用的技术有超声提取(USE)[8,11-14]和加速溶剂萃取(ASE)[6-7,10,15-16]。本实验比较了 USE 和 ASE 法对污泥中 50种目标药物的提取效果。HLB小柱是兼具亲脂和亲水基团的反相柱,可同时富集不同极性的物质,常用于复杂基质中多种目标化合物的同时富集。但对于污泥基质来说,单一的HLB小柱不能较好地去除天然有机质(如腐植酸等)的干扰,从而影响检测结果的灵敏度和特异性[17]。本实验使用HLB串联NH2柱富集净化,结合超高效液相色谱-串联质谱(UPLC-ESI-MS/MS)技术,建立了污泥中多种抗生素的检测方法。
1 实验部分
1.1 仪器与试剂
ACQUITYTM超高效液相色谱仪、Micromass-Quattro UltimaTMPt质谱仪(Waters公司),AllegraTMX-22R型离心机(Beckman公司),超声波清洗器(美国Cole-Parmer公司),Oasis HLB固相萃取柱(6 mL/150 mg)、NH2固相萃取柱(6 mL/500 mg,Waters公司)。
甲醇、乙腈(色谱纯,Fisher Scientific),甲酸(纯度为99%,Acros Organics),氨水、盐酸(优级纯,北京化学试剂公司),乙二胺四乙酸二钠(EDTA-Na2,分析纯,北京化学试剂公司);实验室用水均为超纯水,电阻率为18.2 MΩ·cm(Millipore超纯水机制备)。
6种四环素类和16种喹诺酮类药物购自Sigma公司(St.Louis,MO,USA),20种磺胺类、7种大环内酯类药物和氯霉素购自Dr.Ehrenstorfer公司(Augsburg,Germany),纯度均高于97%。药物名称见表1。
1.2 标准溶液配制
以甲醇为溶剂分别配制质量浓度为1 000 mg/L的标准储备液,于-18℃保存。临用时用5%甲醇水溶液稀释上述标准储备溶液,配制成不同浓度的标准工作液。
1.3 样品采集
污泥样品于2010年5月采自北京某污水处理厂。活性污泥从曝气池中采集,剩余污泥为压滤后的污泥。将不同的污泥样品分别浓缩、冷冻干燥后,于研钵中磨碎并混合均匀,转移至密闭玻璃瓶中于-20℃保存。
1.4 样品前处理
1.4.1 提取准确称取0.5 g污泥,置于50 mL离心管中。加入10 mL乙腈-水(25∶75)溶液(用氨水调节至pH 10.0),涡旋1 min,超声提取15 min后,4℃离心10 min(10 000 r/min)。取出上清液,残渣再加入10 mL提取溶液。重复上述步骤。合并提取液,用水稀释至200 mL。实验考察了ASE的提取效率,称取0.5 g污泥和2.0 g硅藻土充分混匀,全部转移至萃取池中。选取不同的提取溶剂进行提取效率的比较。提取参数:压力1 500 psi,温度100℃,加热时间5 min,静态萃取时间5 min,循环次数3次。转出提取液用水稀释至200 mL。
1.4.2 净化向稀释液中加入0.5 g EDTA-Na2,用盐酸调节稀释液pH值至3.0。将稀释液全部过HLB小柱(活化:6 mL甲醇、6 mL 5%pH 3.0的EDTA-Na2水溶液),上样完毕后,用5 mL水淋洗。再用5 mL甲醇溶液洗脱,洗脱液流经NH2柱(活化:6 mL丙酮),分别用6 mL丙酮-甲醇-甲酸(500∶500∶1)溶液和6 mL丙酮-甲酸(1 000∶1)溶液洗脱。收集洗脱液于微弱的氮气流下吹干,用1 mL 5%甲醇水溶液溶解,涡旋1 min,转至2 mL样品瓶中,用于UPLC-MS/MS测定。
1.5 LC-MS/MS测定
1.5.1 色谱条件 色谱柱:Waters ACQUITY UPLCTMBEH C18柱(100 mm×2.1 mm i.d.,1.7 μm,Waters,USA),柱温:40℃,样品温度:10℃,进样体积:10 μL,流速为0.3 mL/min。
正离子模式:流动相:A为0.1%甲酸的水,B为甲醇;梯度洗脱程序:0~3 min,5%~20%B;3~4 min,20%~30%B;4~8 min,30%~35%B;8~12 min,35%~70%B;12~15 min,70%~100%B;16~16.1 min,100%~5%B;保持3 min。
负离子模式:流动相:A为水,B为乙腈,梯度洗脱程序:0~2 min,5%~45%B;2~5 min,45%~65%B;5~5.1 min,65%~100%B;5.6~5.7 min,100%~5%B;保持3 min。
1.5.2 质谱条件 离子源:电喷雾电离ESI(+)和电喷雾电离ESI(-);毛细管电压:3.0 kV(ESI+)和2.8 kV(ESI-);锥孔电压:60 V;射频透镜1(RF Lens1)和射频透镜2(RF Lens2)电压分别为25 V和1 V;离子源温度:100℃;脱溶剂气温度:350℃;脱溶剂气流量:600 L/min;碰撞室电压:3.4×10-3mbar;50种药物的多反应监测(MRM)参数见文献[5]和[18]。
2 结果与讨论
2.1 前处理条件优化
由于50种目标药物的理化性质差异较大,因此,在优化提取溶液时,使用加标污泥样品比较了不同pH值、不同比例的甲醇-水和乙腈-水,以及USE和ASE两种提取方式的提取效率。结果表明,使用不同比例及组分的提取溶液时,两种提取方式对磺胺类和喹诺酮类药物的提取效率相似;对于四环素类、大环内酯类和氯霉素,USE的提取效率分别为38.2%~77.7%、37.1%~87.4%和40.6%~70.1%;而ASE提取方式只从污泥中提取出泰乐菌素、红霉素和麦迪霉素,提取效率也明显低于USE。这可能是由于四环素类药物比磺胺类药物的热解析温度低[19-20],使用ASE时,温度不能同时满足这几类药物的要求。
采用USE提取,提取液为pH 10.0乙腈-水(25∶75)溶液时,污泥样品中的50种抗生素可以全部提取出来,磺胺类的提取效率为46.1%~69.6%,喹诺酮类为43.1%~70.2%,四环素类为41.9%~71.9%,大环内酯类为65.2%~87.4%,氯霉素为70.1%。明显高于其它提取条件下的提取效率。其原因可能是由于药物在土壤表面的吸附机理包括离子键作用和氢键作用[21-23]。提高提取溶液的pH值可破坏氢键,从而使药物与污泥解离;且在碱性溶液中,喹诺酮类药物结构中的羧基带负电,与活性污泥的负电性表面排斥而达到解离的效果[21]。因此,在碱性条件下的提取效果较好。
2.2 固相萃取条件优化
污泥中的天然有机质(如腐植酸等)含量较高,在液相色谱-质谱分析过程中,会产生基质抑制,造成被测化合物的灵敏度降低。因此,本文采用固相萃取技术对提取液中的目标药物进行富集净化。基于目标药物的极性和酸碱性差异大,本实验比较了HLB柱、MAX柱和C18Sep-Pak柱的富集效果。将提取液稀释至200 mL,加入0.5 g EDTA-Na2并用盐酸调至pH 3.0后,药物在酸性条件下保持非解离状态,其经过HLB柱时可以得到较好的保留[24];其它两种固相萃取柱的上样液均为稀释200 mL的提取液。实验结果表明:药物经过HLB柱后的绝对回收率均高于40%,而MAX柱和C18Sep-Pak柱分别只有24种和29种药物的绝对回收率高于40%。因此,选用HLB柱对污泥样品进行富集。
提取液经HLB富集后,其颜色较为浑浊,基质抑制作用明显,这可能是由于HLB柱不仅吸附目标药物也吸附大量干扰物。当用甲醇洗脱时,由于甲醇的洗脱能力很强,抗生素和干扰物同时被洗出[25]。因此,需对样品净化,本实验选用对腐植酸类杂质净化能力较强的NH2柱。结果表明使用NH2柱净化后,样品提取液颜色澄清,基质抑制率减小为8.6%~25.0%。因此,HLB柱串联NH2柱对污泥样品具有较好的富集净化效果。
2.3 方法学确证
2.3.1 标准工作曲线 50种药物使用外标法定量,配制基质匹配系列标准工作曲线。结果显示,氯霉素标准工作曲线的质量浓度为1.00~160 μg/L;磺胺类和喹诺酮类药物混合标准工作曲线的质量浓度为0.50~100 μg/L;四环素类和大环内酯类药物混合标准工作曲线的质量浓度为5.00~400 μg/L,在相应质量浓度范围内,其线性相关系数均大于0.99。
2.3.2 方法检出限 以能够产生峰对峰信噪比S/N=3和S/N=10对应的药物浓度分别定义为方法检出限(MDL)与方法定量下限(MQL),对于污泥样品中未检出的药物,以基质加标的方法,通过低浓度加标水平的检测结果进行计算。获得目标化合物的MDL为0.03~1.67 μg/kg,MQL为0.10~5.00 μg/kg(见表1)。与已有的文献相比,本方法的检出限处于较低或相当的水平[7-8,16]。
2.3.3 准确度与精密度 用污泥样品进行加标回收实验,设低、中、高3个加标浓度,每个浓度设6个平行。其中低浓度以S0表示,中浓度和高浓度分别为5S0和50S0。S0对应的SDZ、SIM、SMR、SDX、SML、SDM和CAP的浓度为1.0 μg/kg,SMT、SCP、STZ及6种四环素类药物和7种大环内酯类药物的浓度为5.0 μg/kg,其它11种磺胺和16种喹诺酮类药物的浓度为2.0 μg/kg。由于污泥样品中含有某些抗生素,且含量较高,因此对这些抗生素的加标回收实验采用花泥代替,结果见表1。50种目标化合物的平均回收率为40%~111%,相对标准偏差(RSD)为2.9%~27.1%,说明该方法具有较好的回收率和重现性。
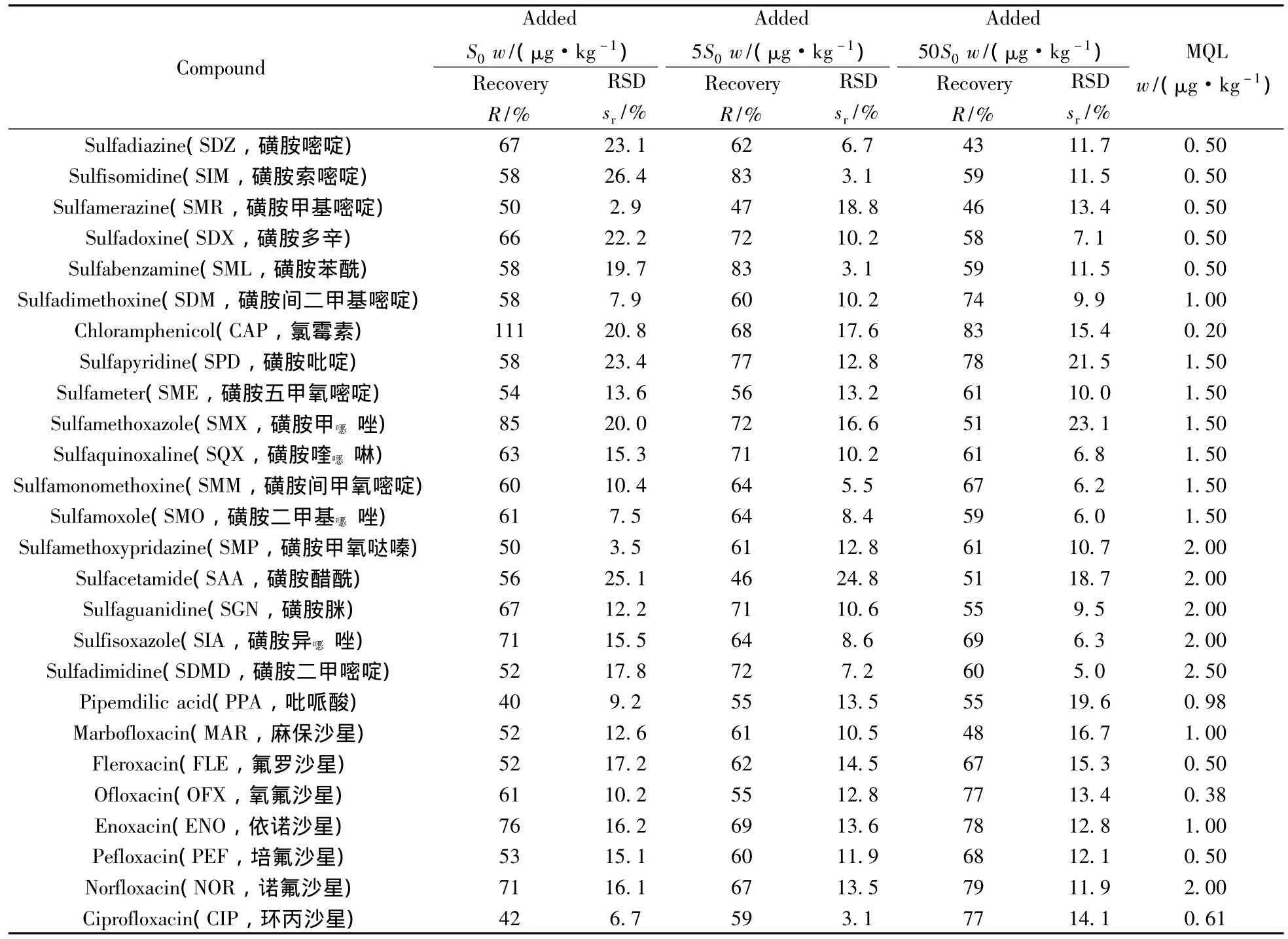
表1 污泥中50种抗生素类药物的回收率(n=6)、相对标准偏差及定量下限Table 1 Recoveries(n=6),relative standard deviations(RSDs)and MQLs of the antibiotics in sludges

(续表1)
2.4 方法应用
应用本方法对北京某污水处理厂采集的6份活性污泥样品和24份剩余污泥样品进行检测,结果见表2,共有14种药物检出,包括3种磺胺类SDZ、SPD和SMX,9种喹诺酮类OFX、NOR、CIP、LOM、ENO、PPA、PEF、ENR和SPA,以及OTC和AZI。其中,磺胺类药物SDZ、SPD和SMX在活性污泥中的浓度水平为39.9~151.1 μg/kg,显著高于剩余污泥中的浓度(4.0~130.2 μg/kg),这可能是由于药物在污泥处理过程中产生降解所致。喹诺酮类药物中NOR、OFX和CIP的检出浓度较高,为57.0~442.5 μg/kg。本实验的检测结果高于或接近其他国家的报道,包括日本(NOR:54~87 μg/kg,CIP:17~243 μg/kg)[13]和爱沙尼亚(NOR:20.8 ~20.5 μg/kg,OFX:4.0 ~10.9 μg/kg 和 CIP:32.8 ~35.5 μg/kg)[7]等。贾瑷等[26]在市政污泥中检出LOM(0.06±0.98 mg/kg)、PPA(0.04±0.27 mg/kg)、SPA(0.01±0.03 mg/kg)和ENR(0.02±0.07 mg/kg)。PEF和ENO只有在水体介质中被检出的报道,在污泥中的报道很少[5,27-28],本实验中这2 种药物的检出浓度分别为9.7 ~34.4 μg/kg 和 51.9 ~174.5 μg/kg。污泥中 OTC的检出浓度与文献报道值相当,如日本(OTC:34 μg/kg)[13]。而污泥中AZI的浓度明显高于德国和瑞士的文献报道(47~158 μg/kg)[16]。图1为标准样品与实际污泥样品的MRM色谱图。

表2 污水处理厂活性污泥和剩余污泥样品中抗生素的检出浓度Table 2 Concentrations of antibiotics in the actived sludge and excess sluge from sewage treatment plants

(续表2)

图1 药物标准样品(A)与实际污泥样品(B)的MRM色谱图Fig.1 MRM chromatograms of the drugs standard solution(A)and sludge sample(B)
3 结论
本研究建立了污泥中氯霉素、磺胺类、喹诺酮类、四环素类和大环内酯类抗生素残留的超高效液相色谱-串联质谱测定方法。使用超声萃取对目标药物进行提取、HLB柱富集、NH2柱净化。该方法便于操作,灵敏度高,可同时测定多种抗生素,满足污泥中污染物痕量分析的要求。
[1]Bai Y.The Antibiotics Abuse Is Sstill the Persistent Ailment(白易.抗生素滥用仍是“顽症”).[2008-11-12].http://www.yybnet.com/www/news/19/63289.html.
[2]Ye Z Q,Weinber H S.Anal.Chem.,2007,79(3):1135 -1144.
[3]Benotti M J,Trenholm R A,Vanderford B J,Holady J C,Stanford B D,Snyder S A.Environ.Sci.Technol.,2009,43(3):597-603.
[4]Choi K J,Kim S G,Kim C W,Kim S H.Chemosphere,2007,66(6):977 -984.
[5]Shao B,Chen D,Zhang J,Wu Y N,Sun C J.J.Chromatogr.A,2009,1216(47):8312 -8318.
[6]Stoob K,Singer H P,Stettler S,Hartmann N,Mueller S R,Stamm C H.J.Chromatogr.A,2006,1128(1/2):1-9.
[7]Lillenberg M,Yurchenko S,Kipper K,Herodes K,Pihl V,Sepp K,L hmus R,Nei L.J.Chromatogr.A,2009,1216(32):5949-5954.
[8]Tang C M,Huang Q X,Yu Y Y,Peng X Z.Anal.Chem.(唐才明,黄秋鑫,余以义,彭先芝.分析化学),2009,37(8):1119-1124.
[9]Yang J F,Ying G G,Zhao J L,Tao R,Su H C,Chen F.Sci.Total Environ.,2010,408(16):3424 -3432.
[10]Golet E M,Strehler A,Alder A C,Giger W.Anal.Chem.,2002,74(21):5455-5462.
[11]Lindberg R H,Wennberg P,Johansson M I,Tysklind M,Andersson B A V.Environ.Sci.Technol.,2005,39(10):3421-3429.
[12]Esther T,Antonio M E,José L T.Anal.Chim.Acta,2006,562:30 -35.
[13]Takashi O,Naoyuki Y,Hiroaki T,Matsukawa H,Tanabe K.Environ.Int.,2009,35:815-820.
[14]Pan X,Ben W W,Qiang Z M.J.Instrum.Anal.(潘寻,贲伟伟,强志民.分析测试学报),2011,30(4):448-452.
[15]Díaz-Cruz M S,Alda M J L,Barceló D.J.Chromatogr.A,2006,1130(1):72 -82.
[16]G bel A,Thomsen A,McArdell C S,Alder A C,Giger W,Theiβ N,L ffler D,Temes T A.J.Chromatogr.A,2005,1085(2):179-189.
[17]Blackwell P A,Lützh ft H C H,Ma H P,Halling-S rensen B,Boxall A B A,Kay P.J.Chromatogr.A,2004,1045(1/2):111-117.
[18]Wang S,Zhang J,Yang Y,Zhang X X,Shao B.Health Res.(王硕,张晶,杨奕,张晓曦,邵兵.卫生研究),2012,41(2):140-146.
[19]Chen R P,Zhang L,Yu J,Tao Y,Zhang Z P,Li K X,Liu D F.Environ.Sci.(陈瑞萍,张丽,于洁,陶芸,张忠品,李克勋,刘东方.环境科学),2012,33(1):156-162.
[20]Jacobsen A M,Halling-Srensen B,Ingerslev F,Hansen S H.J.Chromatogr.A,2004,1038(1/2):157-170.
[21]G bel A,Thomsen A,McArdell C S,Joss A,Giger W.Environ.Sci.Technol.,2005,39(11):3981-3989.
[23]Sun H Y,Shi X,Mao J D,Zhu D Q.Environ.Toxicol.Chem.,2010,29(9):1934-1942.
[24]Ye Z Q,Weinberg H S,Meyer M T.Anal.Chem.,2007,79(3):1135-1144.
[25]Liu H,Zhang G P,Liu C Q.Anal.Chem.(刘虹,张国平,刘丛强.分析化学),2007,35(3):315-319.
[26]Jia A,Wan Y,Xiao Y,Hu J Y.Water Res.,2012,46(2):387-394.
[27]Golet E M,Xifra I,Siegrist H,Alder A C,Giger W.Environ.Sci.Technol.,2003,37:3243 -3249.
[28]Fick J,Soderstrom H,Linberg R H,Phan C,Tvsklind M,Larsson D G J.Environ.Toxicol.Chem.,2009,28(12):2522-2527.
