反反相色谱-串联质谱法直接测定植物源性食品中草甘膦及其代谢物残留
2013-11-28徐敦明林立毅陈鹭平杨黎忠
周 爽,徐敦明,*,林立毅,陈鹭平,周 昱,杨黎忠
(1.厦门出入境检验检疫局 检验检疫技术中心,福建 厦门 361026;2.集美大学 生物工程学院,福建 厦门 361021)
草甘膦(GLY,结构式见图1A)是一种常用的、兼具内吸、传导性、灭生性的除草剂,由于不具有选择性,可有效防除多种杂草及灌木,已被广泛用于农田、果园、道路、林业等,是目前世界上应用最广、生产量最大的广谱性除草剂。虽然GLY属于低毒类的有机磷除草剂,但其不合理使用会导致某些农产品受损(如甘蔗[1]等),影响农产品的国际贸易。此外,植物产品中的GLY残留超标,会影响消费者食用安全,甚至致病、中毒死亡[2-3]。
不同国家(地区)和相关机构对植物(食品)中GLY的限量标准不同:国际食品法典委员会制定了GLY 28项残留限量标准,限量范围在0.05~500 mg之间;美国GLY共172项残留限量标准,限量范围在0.05~400 mg之间;欧盟GLY共340项残留限量标准,其中大豆(干)20 mg、豆类蔬菜0.1 mg等;日本GLY共205项残留限量标准,限量在0.05~20 mg之间。中国已制定了GLY 11项残留限量标准,限量在0.05~6.0 mg之间。
由于GLY和氨甲基膦酸(AMPA)(图1)均为强极性化合物,不溶于大部分有机溶剂,既无紫外吸收,又不易挥发,因此用气相色谱或高效液相色谱检测往往需要借助于柱前或柱后衍生化[4-8],其步骤繁琐,重复性差;而采用离子色谱法时[9-11],由于GLY保留很强,分离时必须采用强淋洗剂碳酸钠,而其它常见阴离子保留较弱无法对GLY和其它阴离子进行同时分析,且仅限于对水等较为单一的基体的测试。近来,用液相色谱-质谱联用技术检测草甘膦及其代谢物的方法也有文献报道,Chen等[12]通过离子色谱与电感耦合等离子体形成八极杆反应池,建立了直接测定土壤中GLY及AMPA的方法,无需衍生化,但操作过程中不仅对缓冲液的pH值要求严格,而且需要注入氦气;Yoshioka等[13]建立了用亲水柱HILIC直接测试人体血清中GLY及AMPA的方法,由于血清基质较为单一,经稀释过滤即可上机分析,前处理方法不适用于食品类复杂基质;Martins-Junior等[14]建立了直接测试黄豆和水中GLY及AMPA的方法,但其检测低限分别为0.3、0.34 mg/kg,达不到痕量分析的要求;Hao等[15]建立了直接测试水中草甘膦及其代谢物的方法,几乎不需前处理即可上机分析;江燕等[16]最近也研究了稻米中GLY及AMPA的测试方法,但其前处理较为复杂,分析时间长达25 min。基于上述方法的局限性,建立一种快速、简捷检测食品中草甘膦及其代谢物的方法十分必要。本文采用去离子水提取蔬菜、水果和茶叶中的GLY及AMPA,用MAX小柱进行净化,同时研究了不同净化条件、流动相以及质谱条件等对测定灵敏度和精密度的影响,前处理方便快捷,不需衍生,在10 min内即可完成对草甘膦及其代谢物的检测,加标实验和实际样品的分析证实该方法切实可行。

图1 GLY(A)与 AMPA(B)的化学结构式Fig.1 Molecular structures of GLY(A)and AMPA(B)
1 实验部分
1.1 仪器与试剂
AB sciex 5000高效液相色谱-质谱仪(美国AB公司);IKA T18 Basic均质器(德国IKA公司);草甘膦(CAS No.1071-83-6,美国Sigma-Aldrich公司);氨甲基膦酸(CAS No.1066-51-9,美国Chem Service公司);乙腈(色谱纯,美国Tedia公司),其他试剂均为分析纯,实验用水为去离子水。
分别准确称取GLY及AMPA各10 mg(精确至0.01 mg),以水为溶剂配制成100 mg/L标准储备液,于4℃保存。实验中根据需要进行稀释。
1.2 分析方法
1.2.1 样品前处理 准确称取样品1 g(不含水)或5 g(含水)至50 mL离心管中,加入10 mL水,均质,放置20 min后,于4 000 r/min离心5 min,取上清液,备用。
1.2.2 样品净化 用3 mL甲醇活化MAX小柱、氨基柱小柱、HLB小柱后,用2 mL水平衡,再加2 mL 2%氨水,弃去。将上述离心后的样品加入小柱内,样品完全附着于小柱内填充物后,加2 mL 2%氨水淋洗,弃去,再依次用2 mL甲醇、2 mL 2%盐酸甲醇洗脱,收集上述洗脱液,氮吹至干,用1 mL水定容后,过0.2 μm滤膜,将滤液取至取样瓶中,待用。
1.2.3 液相色谱条件 色谱柱:Diamond Hydride柱(150 mm×2.1 mm,4.0 μm i.d.);流速:0.8 mL/min;进样量:5 μL;柱温:35℃;流动相:A为5 mmol/L乙酸铵;B为5%水-95%乙腈的乙酸铵溶液(其中乙酸铵浓度为5 mmol/L);梯度洗脱步骤为:0~3 min内,A保持30%,3.1~7 min,A保持95%,7.1~10 min,A保持30%。
1.2.4 质谱条件 扫描方式:电喷雾负离子(ESI-)扫描;检测方式:多反应监测(MRM)模式;雾化气:15 L/min;气帘气:12 L/min;辅助加热气:氮气8 L/min;加热温度:600℃;喷雾电压:5 000 V;定量和定性离子、碰撞能量等参数见表1。

表1 GLY和AMPA的质谱检测参数Table 1 Parameters of MS/MS for GLY and AMPA
1.3 加标回收实验
准确添加标准溶液于空白样品中使样品中GLY浓度为0.02、0.05、0.5 mg/kg,AMPA为0.04、0.1、0.1 mg/kg,室温涡漩振荡5 min,备用。按“1.2”分析方法操作,测其回收率,每个浓度设6次重复,计算样本总体标准偏差和相对标准偏差。
2 结果与讨论
2.1 质谱条件的确立
在电喷雾负离子检测方式下对GLY和AMPA进行一级质谱分析Q1扫描,得到分子离子(即母离子);对分析化合物的分子离子进行二级质谱分析(子离子扫描),得到碎片离子信息,即子离子(图2);然后优化GLY和AMPA的二级质谱参数,以分子离子与特征碎片离子产生的离子对强度达到最大时为最佳,得到每种化合物的二级质谱图;按照二级质谱图提供的碎片离子信息,选择两种化合物的定性离子对(GLY:168.3/63.0;AMPA:110.0/62.9)和定量离子对(GLY:168.3/81.0,168.3/124.0;AMPA:110.0/78.9,110.0/81.0)。
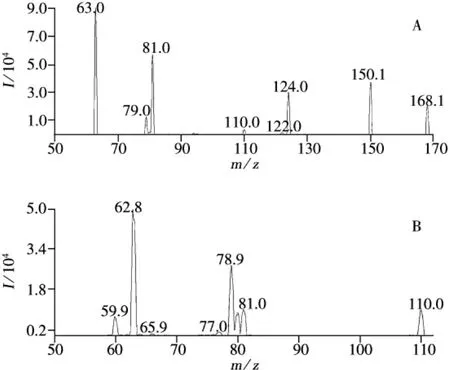
图2 GLY(A)和AMPA(B)的二级离子碎片扫描图Fig.2 Mass spectra of GLY(A)and AMPA(B)in product ion scan
2.2 色谱条件的选择
由于GLY和AMPA的强极性和水溶性,极难在普通反相色谱柱上保留,因此本文比较了两种亲水分离色谱柱HILIC柱和反反相色谱柱的效果。
HILIC亲水色谱柱是一种可以保留和分离极性-离子化合物的色谱柱,但只能在有机相较低的条件下解决极性-离子型化合物的保留问题,而反反相色谱柱同时具有反相色谱机理和正相色谱机理,只需改变流动相即可在反相色谱与正相水平间进行转换,不需要从流动相中完全去除水;使用100%水相洗脱也不会产生柱子崩塌;1 min内可以完成梯度洗脱的再平衡[17-21]。此外,水分子会在HILIC固定相表面形成水膜,而反反相固定相因表面的疏水性较强不会形成水膜。因此,反反相柱不需要用水进行清洗。
鉴于反反相色谱柱的上述优点,本文采用Diamond Hydride反反相色谱柱,对比了不同流动相对色谱的分离和灵敏度。首先考察了不同pH值流动相对色谱分离的影响,结果表明,不同pH值的流动相对GLY及其代谢物的保留几乎无影响;对比了氨水和乙酸铵水溶液对色谱分离的影响,结果表明,乙酸铵水溶液的色谱峰形更符合色谱分析的要求;对比了不同浓度乙酸铵水溶液对色谱分离的影响,发现随着乙酸铵浓度的增加,GLY及其代谢物的保留增强,但超过5 mmol/L后呈下降趋势。因此,本文采用如“1.2.3”所示的流动相,其色谱分离结果如图3所示。

图3 50 μg/L GLY(A)和 50 μg/L AMPA(B)的MRM色谱图Fig.3 Chromatograms of 50 μg/L GLY(A)and 50 μg/L AMPA(B)under MRM mode
2.3 样品净化条件的优化
GLY化学名为N-(膦酰基甲基)甘氨酸,属于弱有机酸,有3个羟基和1个羧基,存在四级电离,分别为:pKa1=0.78,pKa2=2.09,pKa3=5.96,pKa4=10.98;AMPA比GLY少1个羧基,也存在三级电离,因此,GLY和AMPA与溶剂之间既存在离子作用力,又存在氢键的范德华力。本文采用阴离子交换柱MAX小柱、亲水亲酯平衡的HLB小柱和具有强极性的氨基柱,并根据各种小柱的特点和使用条件,对样品进行净化,其净化结果见图4。结果显示,阴离子交换柱MAX的分离效果最好,空白加标回收率在90%以上,HLB柱次之,氨基柱最差。因此本文使用MAX小柱净化样品。

图4 不同净化小柱在不同加标浓度下的净化结果Fig.4 The purification results of different columns spiked concentration(1 -3):20,50,100 μg/L
2.4 方法的线性范围及灵敏度
用标准溶液配制成系列浓度的工作液,GLY的质量浓度为10~500 μg/L,AMPA的质量浓度为20~1 000 μg/L,在优化实验条件下进样(5 μL),以进样质量浓度(X,μg/L)为横坐标,定量离子对的峰面积(Y)为纵坐标建立标准曲线(见表2)。由表2可见,各分析物的相关系数(r2)均大于0.99,相关性良好。
采用向空白样品中逐级降低加标浓度的方法来确定检出限(LOD)和定量下限(LOQ)。以3倍信噪比(S/N=3)对应的目标物浓度作为检出限,以10倍信噪比(S/N=10)对应的目标物浓度作为定量下限,结果如表2所示。

表2 GLY和AMPA的线性方程、检出限(LOD)及定量下限(LOQ)Table 2 Calibration curves,limits of detection(LODs)and limits of quantitation(LOQs)for GLY and AMPA
2.5 方法的准确度、精密度及特异性
在优化条件下,对红茶、绿茶、花茶、青枣、白萝卜、毛豆6种样品进行加标回收实验以及特异性分析。从空白谱图及加标谱图看,分析目标物与基体杂质分离很好,不受基体杂质干扰,加标色谱图和标准溶液色谱图一致(见图5)。每个浓度平行做6个重复,加标回收率和精密度结果见表3。6种基质中GLY、AMPA的回收率为78%~113%,相对标准偏差为3.2%~11.0%。实验结果显示本方法的基质干扰小,重现性和精密度均可满足草甘膦及其代谢物的残留分析要求。

图5 空白红茶样品(A)及其加标样品(B)的色谱图Fig.5 Chromatograms of black tea(A)and spiked blank sample(B)spiked concentration of GLY and AMPA were 0.02 mg/kg and 0.04 mg/kg,respectively

表3 不同基体中GLY和AMPA的加标回收率与相对标准偏差(n=6)Table 3 Recoveries and precisions of GLY and AMPA spiked in the 6 matrixes(n=6)
2.6 实际样品的检测
利用本文建立的分析方法,对100个茶叶样品、50个水果样品和20个蔬菜样品进行测定,有26个样品检出GLY,其中茶叶的检出率最高,检出浓度为0.04~3.0 mg/kg,水果和蔬菜的检出率较低,检出浓度为0.01~0.5 mg/kg。
3 结论
本文采用水提取,MAX小柱净化,反反相液相色谱-串联质谱仪测定,建立了快速检测茶叶、水果、蔬菜等食品中GLY和AMPA的分析方法。利用建立的方法对170个样品进行分析,检出率为21.1%。方法的灵敏度高,回收率良好,适用于食品中GLY和AMPA的测定。
[1]Edward P,Richard J R.Weed Sci.,1991,39:73 -77.
[2]Liang J Z,Liao P J,Li C L.Mod.Prevent.Med.(梁建中,廖培军,黎昌烈.现代预防医学),2003,30(6):900-901.
[3]Zhang D.J.Forensic Med.(张东.法医学杂志),2011,27(3):234-235.
[4]Bernal J,Bernala J L,Martina M T,Nozala M J,Anadónc A,Martínez-Larra aga M R,Martínez M A.J.Chromatogr.B,2010,878:3290-3296.
[5]Cheng X M,Zhou M.Chin.J.Chromatogr.(程雪梅,周敏.色谱),2004,22(3):288.
[6]Qian K,He S,Tang T,Shi T Y,Li J Q,Cao Y S.Food Chem.,2011,127:722 -726.
[7]Li B,Guo D H,Zhu J,Han L,Yang J X.J.Instrum.Anal.(李波,郭德华,朱坚,韩丽,杨景贤.分析测试学报),2005,24:345-348.
[8]Fang F,Xu H,Wei R Q,Liu X N,Xu R,Li S C,Liu B K.J.Instrum.Anal.(方芳,徐会,魏荣卿,刘晓宁,徐蓉,李寿椿,刘宝菎.分析测试学报),2011,30(6):683-686.
[9]Mallat E,Barcelo D.J.Chromatogr.A,1998,823:129 -136.
[10]You J,Koropchak J A.J.Chromatogr.A,2003,989:231-238.
[11]Zhu Y,Zhang F F,Tong C L,Liu W P.J.Chromatogr.A,1999,850:297-301.
[12]Chen Z L,He W X,Beer M,Megharaj M,Naidu R.Talanta,2009,78:852-856.
[13]Yoshioka N,Asano M,Kuse A,Mitsuhashi T,Nagasaki Y,Ueno Y.J.Chromatogr.A,2011,1218:675-680.
[14]Martins-Junior H A,Lebre D T,Wang A Y,Pires M A F,Bustillos O V.Rapid Commun.Mass Spectrom.,2009,23:1029-1034.
[15]Hao C Y,Morse D,Morra F,Zhao X M,Yang P,Nunn B.J.Chromatogr.A,2011,1218:5638-5643.
[16]Jiang Y,Cao Z Y,Jia R L,Qi H,Chen M X.Chin.J.Chromatogr.(江燕,曹赵云,贾瑞琳,齐慧,陈铭学.色谱),2012,30(1):39-44.
[17]Hemstrom P,Ingrum K.J.Sep.Sci.,2006,29:1784-1821.
[18]Soback S.J.AOAC Int.,1999,82:1017-1045.
[19]Ba B B,Saux M C.J.Chromatogr.B,2001,764:349-362.
[20]Peru K M,Kuchta S L,Headley J V,Cessna A J.J.Chromatogr.A,2006,1107:152-158.
[21]Pesek J J,Matyska M T.LCGC,2006,24(3):296-303.
