亨廷顿舞蹈病患者mtDNA D环突变及编码区大片段缺失分析
2013-11-20李晓文
姜 楠,周 璐,肖 海,李晓文#
1)郑州大学临床医学系 郑州 450001 2)郑州大学基础医学院细胞生物学与医学遗传学教研室 郑州 450001
mtDNA突变主要引起神经肌肉系统退行性病变[1],可能是亨廷顿舞蹈病(Huntington’s disease,HD)、Parkinson、小脑共济失调等多种神经系统退行性病变发病的早期信号[2-5]。mtDNA D环对mtDNA的复制和转录有重要的控制作用,D环高变区点突变与多种恶性肿瘤、糖尿病、衰老等疾病密切相关[2-5]。作者初步探讨mtDNA D环突变及编码区大片段缺失与HD发生的关系。
1 对象与方法
1.1研究对象两个HD家系均来自河南省,经知情同意后,抽取患者8人、家系内正常者20人和家系外正常者20人外周静脉血各3 mL进行检测。受检者均经IT-15基因检查明确诊断。
1.2mtDNAD环高变区及编码区突变检测采用酚-氯法提取外周血全基因组DNA,进行PCR。引物序列来源于GenBank,见表1、2,均由上海生工生物工程有限公司合成。mtDNA D环高变区扩增体系共50 μL:Mix(含染料)25 μL,上、下游引物各1 μL(终浓度10 μmol/L),模板DNA 5 μL, ddH2O 18 μL。mtDNA编码区反应体系共20 μL:Mix(含染料)10 μL,上、下游引物各1 μL(终浓度10 μmol/L), 模板DNA 2 μL,ddH2O 6 μL。PCR反应条件均为:95 ℃预变性5 min;94 ℃变性1 min,55 ℃退火1 min,72 ℃延伸35 s,循环35次;72 ℃延伸10 min。产物经20 g/L琼脂糖凝胶电泳,采用凝胶成像分析系统摄影并保存图像。扩增产物由北京金唯智生物公司测序后,与人类线粒体剑桥序列(Cambridge reference sequence,CRS)进行比对。
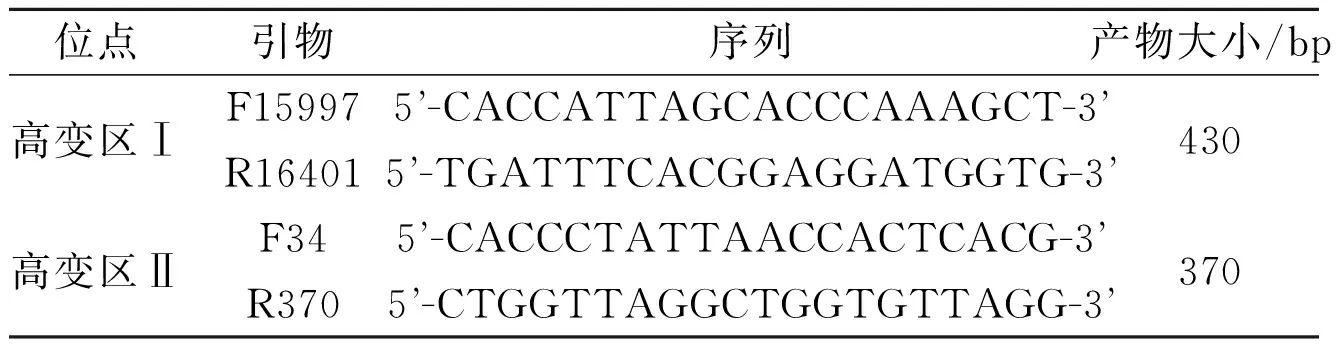
表1 高变区引物序列

表2 编码区引物
2 结果
2.1mtDNAD环高变区检测结果所有测试对象样本均有效扩增。D环高变区Ⅰ、Ⅱ均扩增出相应产物片段,未见片段缺失或插入突变;测序结果与CRS比对,亦未发现片段缺失或插入突变。受检者mtDNA D环高变区测序结果与CRS比对结果见表3~5。其中高变区Ⅰ的rs16061~rs16171是核苷酸歧变的集中区域,突变率达55.6%。
2.2mtDNA编码区检测结果3名(37.5%)患者和16名(80.0%)家系内正常人mtDNA编码区A、B、C、D片段缺失,与CRS比对分析,缺失类型、位置及缺失片段长度见表6。

表3 家系外正常人mtDNA D环高变区测序结果与CRS的比对(n=20)

表4 家系内正常人mtDNA D环高变区测序结果与CRS的比对(n=20)
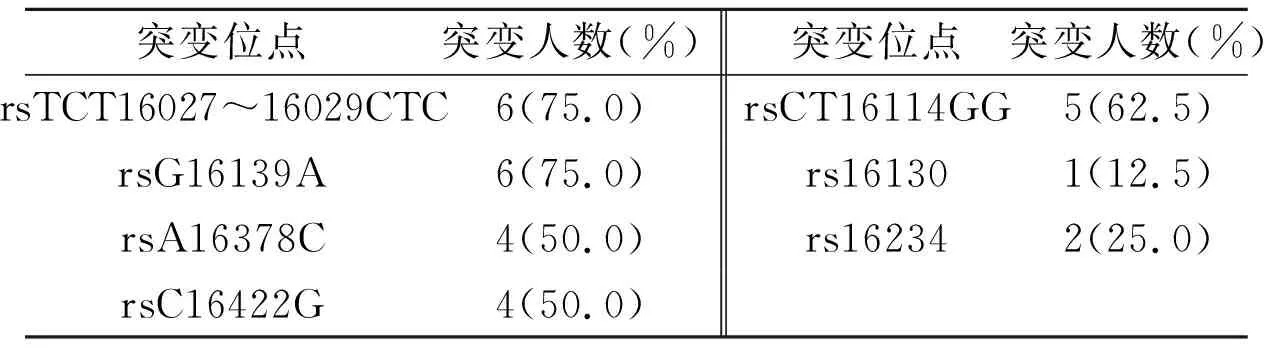
表5 患者mtDNA D环高变区测序结果与CRS的比对(n=8)

表6 mtDNA编码区大片段缺失类型、位置及缺失片段长度
3 讨论
HD是一种迟发型神经系统退行性疾病,病因主要是由于IT-15基因CAG重复序列异常扩增,导致其所编码的亨廷顿蛋白结构与功能异常[6]。线粒体是真核细胞中能量传导的细胞器,参与调控细胞凋亡、基因表达和信号传导,mtDNA突变、氧化磷酸化异常及能量代谢障碍均影响细胞的正常结构及功能。多种神经系统退行性病变中,中枢神经系统糖代谢异常和氧化磷酸化系统损伤出现于神经元丢失、认知功能减退等病理改变之前[4],说明线粒体功能障碍较早参与了多种神经退行性疾病的发病过程。
人类mtDNA是一个双链闭环DNA分子,含有37个基因,编码13种蛋白质、22种转移RNA(tRNA)和2种核糖体RNA(rRNA)[7]。 mtDNA不含内含子,惟一的非编码区是约1 000 bp的D环,又称调控区,是线粒体基因组和核基因组信息交换的枢纽,包含了1个复制起始点、2个转录启动子和4个保守序列,对mtDNA的复制和转录有重要的控制作用。mtDNA具有高突变率且尤以D环区最为常见,包括点突变、片段缺失和插入突变,其16024~16365 nt及73~340 nt两个区域为多态性高发区,分别称为高变区Ⅰ和高变区Ⅱ。作者对HD患者、家系内正常人和家系外正常人的mtDNA D环进行突变检测,未检测到片段缺失或插入,提示mtDNA D环片段重组可能不是HD发病机制中的重要因素。但在rs16139、rs16378、rs16422等位点出现单个碱基改变,提示这些位点可能与HD的易患性或病程演变有关,有待进一步扩大相同母系人员样本证实。此外,与CRS相比,家系外正常人mtDNA D环高变区Ⅰ变异率可达80%,而且变异类型较多,其中约55.6%发生在rs16061~rs16171区域,该区域是核苷酸歧变的集中区域。该结果提示中国汉族人的mtDNA序列与CRS可能存在较明显的种族差异,应进一步扩大样本研究[8]。
有报道[9],约62.5% HD患者mtDNA编码区存在大片段缺失,缺失类型与CAG三核苷酸重复无关,正常人中未见大片段缺失。作者对8名患者及20名家系内正常人进行mtDNA编码区检测,发现3名HD患者和16名家系内正常人存在大片段缺失,未发现HD与mtDNA编码区大片段缺失有特异相关性,推测与mtDNA对表型的阈值效应有关,即家系内正常个体IT-15基因正常,而缺失型mtDNA的数量未能达到阈值,因此不表现出明显的能量代谢障碍。正常个体出现mtDNA编码区大片段缺失的机制有待进一步研究。
[1]Lemasters JJ.Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction,and aging[J].Rejuvenation Res, 2005,8(1):3
[2]Wallace DC.Mitochondrial diseases in man and mouse[J].Science,1999,283(5407):1482
[3]张均田.脑缺血、葡萄糖/能量代谢障碍与神经退行性疾病[J].中国药理学通报,2000,16(3):241
[4]Bubber P,Haroutunian V,Fisch G,et al.Mitochondrial abnormalities in Alzheimer brain, mechanistic implications[J].Ann Neurol,2005,57(5):695
[5]Houshmand M,Panahi MS,Nafisi S,et al.Identification and sizing of GAA trinucleotide repeat expansion, investigation for D-loop variations and mitochondrial deletions in Iranian patients with Friedreich’s ataxia[J].Mitochondrion,2006,6(2):82
[6]Anderson S,Bankier AT,Barrell BG,et al.Sequence and organization of the human mitochondrial genome[J].Nature,198l,290(5806):457
[7]陈伟,陈峰,薛亚丽,等.中国4个民族mtDNA D环多态性研究[J].人类学学报,2000,19(2):127
[8]Myers RH,MacDonald ME,Koroshetz WJ,et al.De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease[J].Nat Genet,1993,5(2):168
[9]Hagelberg E.Recombination or mutation rate heterogeneity? Implications for Mitochondrial Eve[J].Trends Genet,2003,19(2):84
