NF1基因新突变及我国首例NF1基因拷贝数目变异报道*
2013-11-07邓伟平王一鸣
邓 佳, 邓伟平, 钟 诚, 胡 彬, 王一鸣△
1型神经纤维瘤(type 1 neurofibromatosis)是最常见的常染色体显性遗传病之一,在人群中的发病率为1/2 500~1/3 000[1]。主要临床表现为皮肤及皮下神经纤维瘤、咖啡斑和虹膜错构瘤(Lisch结节)。部分患者可具有面容异常及智力低下[2-3]或并发神经胶质瘤[4-5]、恶性周围神经鞘瘤等[6]。神经纤维瘤1(neurofibromatosis 1,NF1;MIM ID:613113)基因是本病的致病基因,定位于染色体17q11.2,含58个编码外显子,有3种剪接方式,其最长的转录亚型编码2 839个氨基酸的蛋白质,外显率接近100%。现已报道的NF1基因突变类型呈多样化,碱基置换、剪切位点突变、小片段的插入和缺失、大片段的缺失、插入及重复、复杂的重排、拷贝数目变异等均有报道,其中关于拷贝数目变异的报道仅限于非中国人群,我国尚未见这一变异的报道。约50%的病例为自发突变所致[7],是人类基因中突变率最高的基因之一。
近年来随着新一代测序技术的迅猛发展,对每一例肿瘤病人进行测序,获得其致病性突变,从而进行个体化治疗[8],这已成为部分西方国家的肿瘤研究/治疗途径。本研究中,我们对2例病人的NF1基因进行分析检测,发现一例国际上未报道的新突变,并首次在国内报道了另一例患者NF1基因的拷贝数目变异。
材料和方法
1 研究对象
2例1型神经纤维瘤的病人均来自于广东省人民医院皮肤科,他们的父母及50例正常对照的血样也来自于广东省人民医院。本研究得到了研究对象及患者家属的知情同意和中山大学伦理委员会批准。
患者S736出生时即发现躯干、四肢有散发的咖啡斑。5岁开始腹部出现散在的皮肤或皮下神经纤维瘤。大小不一,指压瘤体似海绵状并呈局部凹陷,松开手指即恢复原状,同时伴有色素沉着斑,但一直无自觉症状。随着年龄增长,皮损波及躯干四肢。患者的智力、身体发育均正常,不伴有眼睛及周围神经系统症状。另一患者S743出生时即出现了与患者S736相似的皮肤表现,此外还于幼年期出现了腰椎右侧弯曲,认知能力较差,以致不能完成初中学业并最后缀学,患者于19岁死于脑胶质瘤。按NIH标准[9],两患者均诊断为1型神经纤维瘤,两患者的父母经详细的临床检查未发现异常。
2 方法
2.1 基因组DNA提取 抽取患者及其父母和50例正常对照外周血2 mL,乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)抗凝,按 Qiagen 基因组小量抽提试剂盒指南提取DNA。
2.2 引物设计及PCR 从数据库UCSC Human Genome Browser(http://genome.ucsc.edu)(GRCh37/hg19)提取NF1基因的序列 (NG_009018.1),利用Oligo 6.0(http://www.oligo.net/downloads.html)自行设计能够扩增基因编码区域及剪切位点的引物,并由上海生工生物公司合成。PCR扩增反应体系为30μL标准体系,反应条件:95℃预变性3 min;95℃30 s,退火 45 s,72 ℃ 1 min,35个循环;72 ℃ 10 min。
2.3 PCR产物测序及突变鉴定 对PCR产物进行直接测序,所用试剂为Big Dye Terminator 3.1 Cycle Sequencing Kit(Applied Biosystems),在 ABI 3730XL DNA自动测序仪进行。对测序结果用Sequence Scanner 1.0软件(ABI)分析,所得序列与UCSC数据库进行比对。碱基变异按den Dunnen以及Antonarakis(http://www.hgvs.org/mutnomen/)命 名 法则[10]。对所发现的遗传变异与人类基因突变数据库(the Human Gene Mutation Database,HGMD;http://www.hgmd.org/)中的数据进行比对,并查询PubMed上已发表的文献确定所检突变是否为新突变。突变根据以下参考序列命名:GenBank NM_001042492.2 和 GenBank NP_001035957.1。
2.4 多重连接探针扩增技术 在测序中对于未发现突变的患者S743,我们推测为NF1拷贝数目改变所致,故进一步采用了多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)进行分析。为了排除多重突变的情况,我们对已发现突变的病人S736也进行了此分析。MLPA 1型神经纤维瘤检测试剂盒购自MRC-Holland,外显子区域及边界区域探针包含在以下3组探针盒中:SALSA P081、P082和P122。实验流程按MDA-002版本MLPA说明书进行操作,产物经ABI 3730XL DNA自动测序仪电泳后,对获得的GeneMapper数据用Coffalyser软件进行分析。
2.5 长片段PCR 由于MLPA实验提示患者S743存在NF1基因的大片段缺失,我们根据MLPA检测的结果,结合国际上已报道的NF1 3个常见缺失片段的断裂部分,合成了长片段PCR引物(表1),并进行相应的PCR反应。
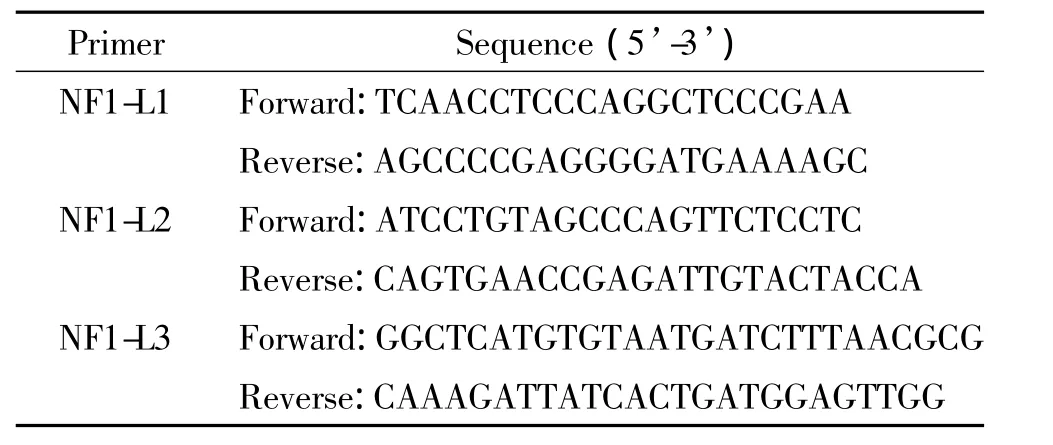
表1 检测断裂点的长片段PCR引物序列Table 1.Primers used for the long range PCR to detect the breakpoint
结 果
1 突变检测结果
正反向测序证实病人S736在NF1基因中存在1 个新突变:c.6345_6346 ins G(p.Leu2116Alafs*4),见图1A。该突变造成开放阅读框架的移位,在肽链第2 119位引入终止密码子,从而使3’端721个氨基酸被截断,丢失了野生型蛋白中犰狳式褶皱(ARM-type fold)结构域C末端的一部分(InterPro,http://www.ebi.ac.uk/interpro/),见图 2。SIFT 预测,此突变还可引起无义突变介导的mRNA降解(nonsense mediated decay,NMD),导致含该突变的等位基因无实际上的表达。此突变经3次以上正反向验证。HGMD检索并未发现此突变的报道,故为一新突变,50例无血缘关系的正常人中未发现同样突变。获此结果后,我们进一步对其父母进行这一突变的检测,未发现异常,见图1B、C,故此突变为一de novo突变。病人S743在NF1基因的编码外显子及其侧翼序列未发现突变。

Figure 1.Partial sequencing results of patient S736(A)and her parents(B,C).Arrows indicate the insertion of G in patient S736 and the wild C in her parents.图1 患者S736及其父母的部分测序图结果
2 MLPA结果
病人S743 MLPA检测结果提示1.3~1.9 Mb缺失(图3),缺失片段涵盖整个NF1基因及其部分5’和3’端的侧翼部分。病人S736 MLPA检测未见异常。
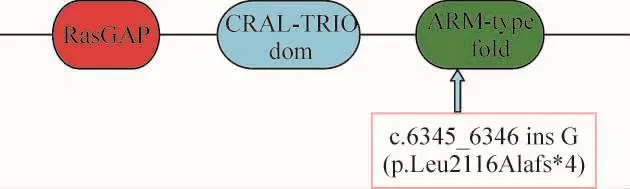
Figure 2.Mutant NF1 protein of patient S736.The wild-type NF1 protein has an armadillo(ARM)-type fold domain(green)and two domains:Ras-GAP(red),CRALTRIO(blue).Arrow denotes the truncating point of the mutation p.Leu2116Alafs*4,which would result in the loss of part of the ARM-type fold.图2 患者S736的突变NF1蛋白

Figure 3.The MLPA results showed a 1.3 ~1.9 Mb deletion in patient S743.A:patient S743.Probes with* (red)show the heterozygous deletion positions.B:normal control.图3 患者S743的MLPA结果显示有1.3~1.9 Mb缺失
3 长片段PCR
长片段PCR未获得任何特异性扩增片段,提示这一病例的缺失可能存在不同于以往报道的断裂点。
讨 论
本研究先对2例1型神经纤维瘤患者进行了NF1基因的突变筛查。通过对PCR产物直接测序的方法,在患者S736中发现了NF1基因新生突变c.6345_6346 ins G(p.Leu2116Alafs*4),HGMD 检索证明该突变为一个新突变。此后,对2个病人进行了MLPA检测,病人S743检测到整个NF1基因的缺失和5’和3’端侧翼序列的缺失,且缺失区域在1.3~1.9 Mb范围内,文献检索证明这是国内第一次用MLPA对NF1基因进行检测,也是首次中国人整个NF1基因缺失的报道。
p.Leu2116Alafs*4使NF1蛋白的第2 116位氨基酸由亮氨酸变成丙氨酸,并且由于碱基的插入导致了密码子的框移,在突变位点后的第3个密码子处出现终止密码子从而使蛋白质结构截断,p.Leu2116Alafs*4位于 ARM-type fold结构域。ARM-type fold是一个多超螺旋褶皱,由2弧层α螺旋排列在一个常规的右手超螺旋中,这种结构提供了一个广泛的溶剂表面,很适合与底物如蛋白质和核酸结合。p.Leu2116Alafs*4突变截断了野生型蛋白质中的部分ARM-type fold结构域(图2),可能影响它的超螺旋结构从而影响NF1蛋白与DNA或蛋白质的结合及相互作用,最终导致功能的丧失;且由于无义突变介导的mRNA降解,此突变不存在转录蛋白,相当于等位基因缺失。该患者父母体格检查正常,皮肤黏膜未发现异常,且突变筛查结果阴性,说明该患者的突变是de novo突变,50例正常对照的筛查也排除了多态性的可能,因此本研究所发现的NF1 de novo突变是引起1型神经纤维瘤的致病性突变。
1型神经纤维瘤病人中,约5%[11]病人存在大片段的NF1基因缺失。患者S743经MLPA检测提示其存在含整个NF1基因的一段1.3~1.9 Mb的缺失,非等位基因同源重组被认为是导致大片段缺失发生的最常见原因,国外研究已发现3个重组热点:1.4 Mb的缺失称为1型,是大片段缺失最常见的一型,断裂点主要位于 NF1-REPs A 和 C[12-13];1.2 Mb的缺失称为2型,断裂点位于SUZ12和它的假基因SUZ12P 内[14];1.0 Mb的缺失称为3 型,断裂点位于NF1-REPs B 和 C[3,15]。大片段的 NF1 缺失经常与严重疾病表型相关,如先天性面部畸形、智力缺陷及对恶性肿瘤易感性高。S743患者于19岁患脑神经胶质瘤而死亡,属重型1型神经纤维瘤患者,这显然与它缺失NF1基因相关。NF1基因是一个肿瘤抑制基因,它能下调原癌基因ras的活性[16]。由于我们针对3个常见断裂点的长片段PCR未获得阳性结果,此患者可能含有新的不常见的断裂点,目前我们对于S743患者的断裂点研究仍在进行中。
[1] Williams VC,Lucas J,Babcock MA,et al.Neurofibromatosis type 1 revisited[J].Pediatrics,2009,123(1):124-133.
[2] Mautner VF,Kluwe L,Friedrich RE,et al.Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions[J].J Med Genet,2010,47(9):623-630.
[3] Pasmant E,Sabbagh A,Spurlock G,et al.NF1 microdeletions in neurofibromatosis type 1:from genotype to phenotype[J].Hum Mutat,2010,31(6):E1506-E1518.
[4] Thiagalingam S,Flaherty M,Billson F,et al.Neurofibromatosis type 1 and optic pathway gliomas:follow-up of 54 patients[J].Ophthalmology,2004,111(3):568-577.
[5] Graf N.Glioblastoma in children with NF1:the need for basic research[J].Pediatr Blood Cancer,2010,54(7):870-871.
[6] McCaughan JA,Holloway SM,Davidson R,et al.Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1 [J].J Med Genet,2007,44(7):463-466.
[7] Lázaro C,Ravella A,Gaona A,et al.Neurofibromatosis type 1 due to germ-line mosaicism in a clinically normal father[J].N Eng J Med,1994,331(21):1403-1407.
[8] Hezel AF,Deshpande V,Zhu AX.Genetics of biliary tract cancers and emerging targeted therapies[J].J Clin Oncol,2010,28(21):3531-3540.
[9] National Institutes of Health Consensus Development Conference.Neurofibromatosis:conference statement[J].Arch Neurol,1988,45(5):575-578.
[10] den Dunnen JT,Antonarakis SE.Mutation nomenclature extensions and suggestions to describe complex mutations:a discussion [J].Hum Mutat,2000,15(1):7-12.
[11] Kluwe L,Siebert R,Gesk S,et al.Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene[J].Hum Mutat,2004,23(2):111-116.
[12] De Raedt T,Stephens M,Heyns I,et al.Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion [J].Nat Genet,2006,38(12):1419-1423.
[13]Forbes SH,Dorschner MO,Le R,et al.Genomic context of paralogous recombination hotspots mediating recurrent NF1 region microdeletion [J].Genes Chrom Cancer,2004,41(1):12-25.
[14] Petek E,Jenne DE,Smolle J,et al.Mitotic recombination mediated by the JJAZ1(KIAA0160)gene causing somatic mosaicism and a new type of constitutional NF1 microdeletion in two children of a mosaic female with only few manifestations[J].J Med Genet,2003,40(7):520-525.
[15] Bengesser K,Cooper DN,Steinmann K,et al.A novel third type of recurrent NF1 microdeletion mediated by nonallelic homologous recombination between LRRC37B-containing low-copy repeats in 17q11.2 [J].Hum Mutat,2010,31(6):742-751.
[16] Trovó-Marqui AB,Tajara EH.Neurofibromin:a general outlook [J].Clin Genet,2006,70(1):1-13.
