RXR激动剂通过抑制PKC激活对抗高糖诱导的大鼠血管平滑肌细胞增殖*
2013-11-07柴大军许昌声宁若冰林金秀
柴大军, 许昌声, 宁若冰, 祝 江, 谢 泓, 林金秀
糖尿病患者发生动脉粥样硬化性疾病的风险较正常人显著增加。血管平滑肌细胞(vascular smooth muscle cells,VSMCs)增殖及其由血管中膜向内皮下区域迁移是动脉粥样硬化形成和发展过程中的重要事件,在血管基底膜退化和重构过程中起到核心作用[1],蛋白激酶 C(protein kinase C,PKC)参与了血管平滑肌细胞的增殖过程[2]。哺乳动物的细胞增殖过程通常由细胞周期所调控,细胞从G1期进入S期需要细胞周期蛋白(cyclin)/细胞周期蛋白依赖激酶(cyclin-dependent kinase,CDK)复合物的结合和激活,主要是 cyclin/CDK4 和 cyclin/CDK2[3]。p27Kip1是Cip/Kip家族的细胞周期蛋白依赖激酶抑制物(cyclin-dependent kinase inhibitors,CDKI),能够与cyclin/CDK4和cyclin/CDK2复合物结合并抑制后者的激酶活性,从而负性调控细胞周期进程[4],研究发现p27Kip1是调控VSMCs增殖的重要因素[5]。视黄醇类X受体(retinoid X receptor,RXR)作为核激素受体超家族的成员,介导了多种激素和药物的细胞生物学效应。RXR在核受体家族中具有独特的地位,因其可以和许多核受体形成异源二聚体并在心血管功能的调控方面具有重要作用[6],在体研究发现RXR激动剂具有抗动脉粥样硬化效应[7]。然而,RXR及其配体通过何种机制来调控动脉粥样硬化等心血管事件的进展尚不清楚。本研究将讨论RXR激动剂在高糖诱导大鼠主动脉平滑肌细胞(rat aortic smooth muscle cells,RASMCs)增殖过程中的作用及相关机制,为RXR及其配体心血管保护作用的发挥寻求新的分子机制。
材料和方法
1 材料
DMEM(Dulbecco’s modified Eagle medium)培养基,胎牛血清(fetal bovine serum,FBS)、胰蛋白酶、磷酸缓冲液、Trizol和 Opti-MEM购自Invitrogen;PKC抑制剂(PKC inhibitor peptide)和BrdU增殖检测试剂盒购自Millipore;II型胶原酶和RXR天然配体9-顺式维甲酸(9-cis-retinoic acid,9-cis-RA)购自Sigma;RXR特异性配体SR11237由厦门大学药学院曾锦章教授友情提供;WST-1细胞增殖检测试剂盒购自Cayman;兔抗鼠β-actin、PKC及 p-PKC单克隆抗体和兔抗鼠CDK2多克隆抗体、羊抗鼠p27Kip1多克隆抗体和辣根过氧化物酶标记的Ⅱ抗购自Santa Cruz。
2 方法
2.1 组织干涸法体外培养RASMCs 在参照文献方法[8]的基础上稍加改进,取10~12周龄健康雄性Sprague-Dawley大鼠的胸主动脉分离培养RASMCs。去内皮化的组织块均匀平铺于0.1%明胶包被的培养瓶内培养10 d。应用平滑肌肌动蛋白抗体进行免疫细胞化学染色方法鉴定细胞,3~6代生长良好的细胞(细胞纯度>95%)用于本研究。
2.2 RASMCs的干预和分组 25 mmol/L高浓度葡萄糖常被用来刺激内皮细胞,体外模拟糖尿病患者的体内高糖环境[9]。实验过程中设立等渗透压的甘露醇干预组(5.5 mmol/L葡萄糖 +19.5 mmol/L甘露醇)作为对照以排除高糖干预时因细胞外渗透压急性改变产生的细胞学效应[10],分为正常组(葡萄糖终浓度为5.5 mmol/L)、甘露醇组、高糖组(葡萄糖终浓度为 25 mmol/L)、高糖 +9-cis-RA(10-9~10-7mol/L)组、高糖 +SR11237(10-7mol/L)组和高糖+PKC抑制剂(2~20 μmol/L)组。各组细胞在干预前用无血清的DMEM培养基培养24 h使细胞进入静止期。
2.3 细胞增殖活性测定 细胞增殖活性检测采用WST-1细胞增殖检测试剂盒,检测方法参考产品说明书进行。
2.4 溴脱氧鸟苷(bromodeoxyuridine,BrdU)插入法检测RASMCs DNA合成 采用BrdU增殖检测试剂盒依据产品说明检测RASMCs DNA合成。细胞干预结束,先用BrdU标记4 h,再与过氧化物酶标记的抗BrdU抗体共孵育。加入底物3-甲基联苯胺并用酶标仪450 nm波长读取吸光度(absorbance,A),间接反映细胞BrdU的掺入率。
2.5 细胞周期分析 参照文献[11],采用流式细胞学方法检测RASMCs细胞周期分布比例。细胞干预后,用70%冰乙醇进行固定,用含有40 mg/L核糖核酸酶的PBS 37℃孵育30 min,再用含50 mg/L碘化丙啶的PBS悬浮细胞。细胞周期分布比例用FACSCalibur Sorting System(Becton& Dickinson)检测,细胞在G0/G1、S和G2/M期的分布百分比采用Modfit LT软件进行分析。
2.6 细胞蛋白的提取和免疫印迹杂交 提取细胞总蛋白,煮沸变性10 min,采用10% ~12%SDS-PAGE电泳分离50 μg细胞蛋白样品,转至PVDF膜后用特异的抗CDK2、p27Kip1及p-PKC抗体进行免疫杂交检测,β-actin作为内参照。
3 统计学处理
数据以均数±标准误(mean±SEM),采用统计软件SPSS 13.0分析处理。组间均数比较采用单因素方差分析(One-Way ANOVA),两两比较采用Scheffe事后检验(Post hoc Scheffe test),以P<0.05为差异有统计学意义。
结 果
1 RXR激动剂抑制高糖诱导的RASMCs增殖
WST-1细胞增殖检测结果显示,高糖(25 mmol/L)干预RASMCs 48 h后,细胞增殖活性较对照组(葡萄糖终浓度为5.5 mmol/L)显著增加(1.46 ±0.10 vs 0.46 ±0.07,P <0.01),等渗透压的甘露醇干预 48 h未增加细胞增殖活性,9-cis-RA呈浓度依赖性(10-9~10-7mol/L)地抑制高糖环境下RASMCs细胞增殖活性的增加幅度,10-7mol/L的SR11237对高糖诱导RASMCs细胞增殖活性的抑制作用与等浓度的9-cis-RA相似,见图1。

Figure 1.RXR agonists inhibited high-glucose-induced proliferation of RASMCs.RASMCs were incubated with 9-cis-RA(10-9~10-7mol/L)or SR11237(10-7mol/L)for 1h and then exposed to high glucose or mannitol for 48 h.RASMCs proliferation was analyzed by WST-1 assay.Mean ± SEM.n=5.*P < 0.05 vs normal glucose;△P<0.05 vs high glucose alone;#P <0.05 vs high glucose plus 10-9mol/L 9-cis-RA;▲P <0.05 vs high glucose plus 10-8mol/L 9-cis-RA.图1 RXR激动剂抑制高糖诱导的RASMCs增殖
2 RXR激动剂抑制高糖诱导的RASMCs DNA合成
9-cis-RA呈浓度依赖性地抑制高糖诱导下RASMCs的DNA合成,10-7mol/L SR11237的抑制效应与10-7mol/L 9-cis-RA相似,见图2。

Figure 2.RXR agonists inhibited high-glucose-induced DNA synthesis in RASMCs.RASMCs were incubated with 9-cis-RA(10-9 ~ 10-7mol/L)or SR11237(10-7 mol/L)for 1 h and then exposed to high glucose or mannitol for 48 h.RASMCs DNA synthesis was detected by BrdU incorporation assay.Mean±SEM.n=5.*P <0.05 vs normal glucose;△P <0.05 vs high glucose alone;#P<0.05 vs high glucose plus 10-9 mol/L 9-cis-RA;▲P <0.05 vs high glucose plus 10-8mol/L 9-cis-RA.图2 RXR激动剂抑制高糖诱导的RASMCs DNA合成
3 RXR激动剂对高糖环境下RASMCs细胞周期进展的影响
无血清DMEM培养基培养RASMCs 24 h后可使(85.6±3.8)%的细胞同步于 G0/G1期,高糖干预48 h使 RASMCs分布于 S期的比例由(1.58±0.08)%增加至(16.28±1.24)%(P<0.05)。当用9-cis-RA(10-9~10-7mol/L)或 SR11237(10-7mol/L)预处理RASMCs 1 h后再给予高糖干预48 h,结果显示9-cis-RA呈浓度依赖性地下调RASMCs在S期的分布比例,10-7mol/L的SR11237对RASMCs在S期分布比例的下调作用与等浓度9-cis-RA的作用相当[分别由(16.28±1.24)%下降至(4.98±0.89)%和(4.18±0.76)%],见图3。
4 RXR激动剂对高糖环境下RASMCs CDK2和p27Kip1蛋白表达的影响
高糖干预48 h后,RASMCs细胞CDK2蛋白的表达显著增加,p27Kip1蛋白水平则明显下降。9-cis-RA呈浓度依赖性地降低高糖诱导下CDK2蛋白表达水平增加的幅度,同时逆转高糖对p27Kip1蛋白的下调作用,10-7mol/L SR11237对高糖环境下CDK2和p27Kip1蛋白表达的影响与10-7mol/L 9-cis-RA相似,见图4。

Figure 3.Effects of RXR agonists on high-glucose-induced cell cycle progression of RASMCs.The cell cycle was determined by flow cytometry.The statistical analysis of five independentexperimentswith similarresults showed the inhibition of 9-cis-RA and SR11237 on high-glucose-induced RASMCs cell cycle progression.Each item is derived from a representative experiment where data from at least 10 000 events were obtained.图3 RXR激动剂抑制高糖诱导的RASMCs细胞周期进程
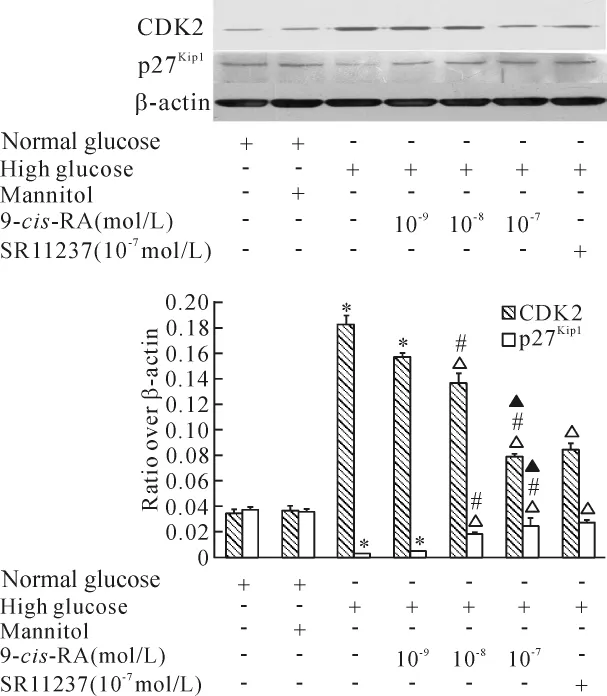
Figure 4.Effects of RXR agonists on the expression of CDK2 and p27Kip1in RASMCs.RASMCs were incubated with 9-cis-RA(10-9 ~10-7mol/L)or SR11237(10-7mol/L)for 1 h and then exposed to high glucose or mannitol for 48 h.Mean±SEM.n=4.*P <0.05 vs normal glucose;△P<0.05 vs high glucose alone;#P<0.05 vs high glucose plus 10-9mol/L 9-cis-RA;▲P <0.05 vs high glucose plus 10 -8mol/L 9-cis-RA.图4 在高糖环境下,RXR激动剂下调CDK2表达,上调p27Kip1表达
5 PKC参与高糖诱导的RASMCs增殖
PKC抑制剂呈浓度依赖性的抑制高糖环境下RASMCs细胞的增殖,见图5A。20 μmol/L的 PKC抑制剂显著抑制高糖诱导的RASMCs DNA合成(图5B)和细胞周期进展过程 (图5C)。因此,PKC参与高糖环境下RASMCs的增殖过程并发挥重要作用。
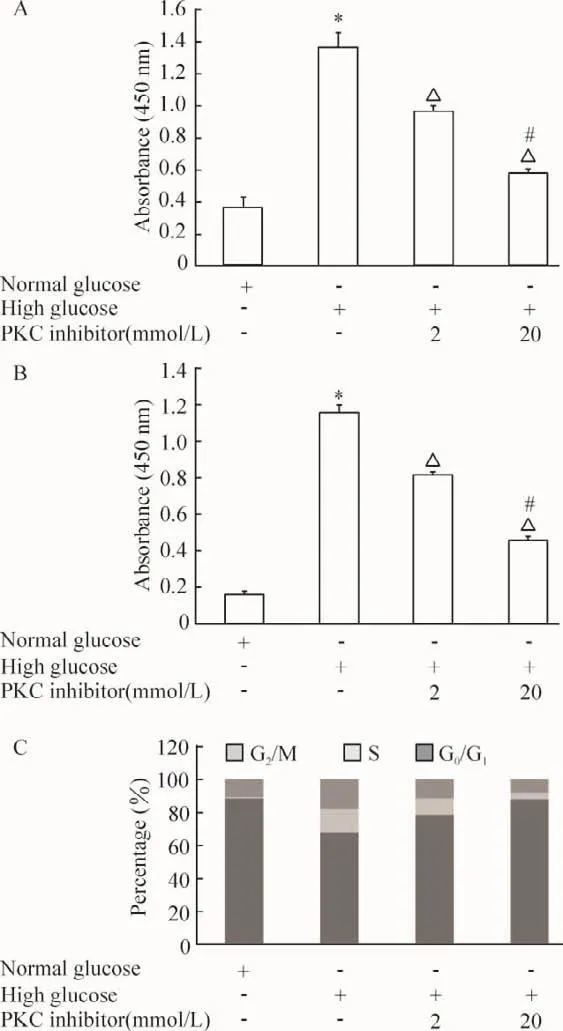
Figure 5.PKC inhibitor inhibited high-glucose-induce RASMCs proliferation.A:RASMCs proliferation was analyzed by WST-1 assay;B:RASMCs DNA synthesis was detected by BrdU incorporation assay;C:flow cytometry analysis of the effect of PKC inhibitor peptide on high-glucose-induced cell cycle progression.RASMCs were treated with the indicated concentration of PKC inhibitor peptide for 1 h and then exposed to high glucose for 48 h.Mean±SEM.n=5.*P <0.05 vs normal glucose;△P<0.05 vs high glucose alone;#P <0.05 vs high glucose plus 2 μmol/L PKC inhibitor.图5 PKC抑制剂抑制高糖环境下RASMCs的增殖
6 PKC参与高糖对RASMCs CDK2和p27Kip1蛋白表达的调控
PKC抑制剂呈浓度依赖性地降低高糖诱导下CDK2蛋白表达水平增加的幅度,同时上调高糖干预下p27Kip1蛋白的表达,见图6。这说明PKC参与了高糖对RASMCs CDK2和p27Kip1蛋白表达的调控。

Figure 6.Effects of PKC inhibitor on the expression of CDK2 and p27Kip1in RASMCs.RASMCs were incubated with PKC inhibitor peptide for 1 h and then exposed to high glucose for 48 h.Mean ± SEM.n=4.*P <0.05 vs normal glucose;△P <0.05 vs high glucose alone;#P <0.05 vs high glucose plus 2 μmol/L PKC inhibitor.图6 PKC抑制剂在高糖环境下调CDK2的表达,上调p27Kip1的表达
7 RXR激动剂抑制高糖诱导的PKC活化
用 9-cis-RA(10-8~ 10-7mol/L)或 SR11237(10-7mol/L)预处理RASMCs 45 min后,再暴露于高糖环境30 min,发现9-cis-RA呈浓度依赖性抑制高糖诱导的PKC磷酸化水平,SR11237与等浓度9-cis-RA具有相似效应,见图7。
讨 论
RXR是由特异性配体激活的转录因子,可与其它核受体(如肝脏X受体和过氧化物酶体增生物激活受体等)形成异源二聚体在调控机体能量代谢、炎症、氧化应激及胶原合成等方面发挥有益作用[12-14]。RXR配体通过减少肠道脂质吸收和降低脂蛋白水平抑制动脉粥样斑块的进展[7],我们前期发现RXR激动剂通过抑制高糖环境下PKC的活化对抗高糖对人血管内皮细胞产生的氧化应激损伤[15],提示RXR可能成为新的抗动脉粥样硬化的药物治疗靶点。
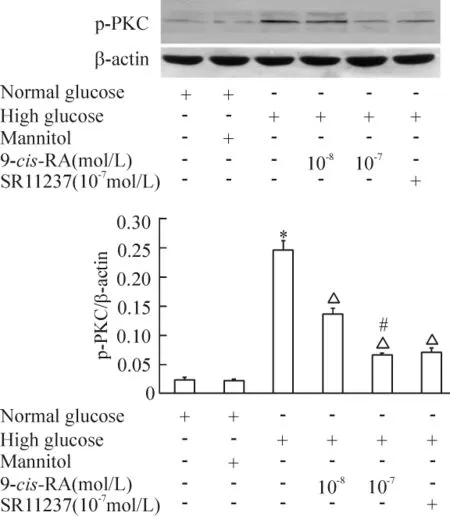
Figure 7.RXR agonists inhibited high-glucose-induced PKC activation.Mean± SEM.n=4.*P <0.05 vs normal glucose;△P<0.05 vs high glucose alone;#P<0.05 vs high glucose plus 10 -8mol/L 9-cis-RA.图7 RXR激动剂抑制高糖诱导的PKC磷酸化
血管平滑肌细胞的增殖和迁移是糖尿病致动脉粥样硬化形成过程中的重要环节,受到细胞周期蛋白、CDK和CDKI等因素的调控[3-4]。本研究通过WST-1细胞增殖分析和DNA合成检测方法发现了RXR天然配体9-cis-RA可显著抑制高糖诱导的RASMCs增殖和DNA合成。在高糖环境下,RASMCs在细胞周期S期的分布比例显著增加,但9-cis-RA可显著逆转高糖对细胞周期的调控作用。9-cis-RA除与RXR结合外,还可与维甲酸受体结合,但RXR的特异性配体SR11237与等浓度9-cis-RA在对抗高糖的促增殖效应方面具有相似作用,说明9-cis-RA的钝化效应是通过RXR特异性通路实现的。CDK2是调节细胞增殖和细胞周期进程的重要细胞周期蛋白依赖激酶,但受到细胞周期蛋白依赖激酶抑制物p27Kip1的调控,后者通过抑制CDK2的活性实现对细胞周期进程的负性调控[3-5]。为深入探讨RXR激动剂在高糖条件下抑制RASMCs增殖的分子机制,我们观察了9-cis-RA和SR11237对CDK2和p27Kip1蛋白的影响,结果发现高糖对CDK2和p27Kip1蛋白表达的作用可被9-cis-RA和 SR11237所逆转。因此,RXR激动剂通过调控CDK2和p27Kip1蛋白的表达对抗高糖的促增殖效应。
PKC作为丝氨酸/苏氨酸激酶家族的成员在细胞信号转导过程中发挥重要作用,参与调控心脏收缩、心肌肥厚、缺血/再灌注损伤、氧化应激反应和动脉粥样硬化等病理生理过程[15-18],但在特定细胞中可被RXR所调控[15]。在本研究中,PKC抑制剂通过上调p27Kip1、下调CDK2蛋白表达抑制高糖诱导的RASMCs增殖,证实了PKC参与高糖环境下血管平滑肌细胞的增殖过程。为揭示RXR激动剂调控CDK2和p27Kip1蛋白表达和抑制血管平滑肌细胞增殖的机制,我们再次探讨了RXR配体对PKC活化的影响,结果显示9-cis-RA和SR11237可钝化高糖诱导的PKC活化。所以,抑制PKC活化是RXR激动剂在高糖环境下调CDK2、上调p27Kip1及最终实现抑制RASMCs增殖的重要机制。维甲酸(retinoid acid,RA)可能通过影响PKC的亚细胞定位或与PKC直接结合钝化PKC[19-20],但RXR配体抑制PKC活化的机制仍需深入探讨。
总之,本研究发现了在高糖环境下RXR激动剂通过抑制PKC的激活实现其对血管平滑肌细胞CDK2和p27Kip1的调控,进而抑制细胞增殖,为RXR及其配体的抗动脉硬化作用探明新的分子机制。
[1] Orr AW,Sanders JM,Bevard M,et al.The subendothelial extracellular matrix modulates NF-κB activation by flow:a potential role in atherosclerosis[J].J Cell Biol,2005,169(1):191-202.
[2] Ding Q,Chai H,Mahmood N,et al.Matrix metalloproteinases modulated by protein kinase Cε mediate resistininduced migration of human coronary artery smooth muscle cells[J].J Vasc Surg,2011,53(4):1044-1051.
[3] Sherr CJ,Roberts JM.CDK inhibitors:positive and negative regulators of G1-phase progression[J].Genes Dev,1999,13(12):1501-1512.
[4] Noori S,Hassan ZM.Tehranolide inhibits proliferation of MCF-7 human breast cancer cells by inducing G0/G1arrest and apoptosis[J].Free Radic Biol Med,2012,52(9):1987-1999.
[5] Tanner FC,Boehm M,Akyurek LM,et al.Differential effects of the cyclin-dependent kinase inhibitors p27Kip1,p21Cip1,and p16Ink4on vascular smooth muscle cell proliferation[J].Circulation,2000,101(17):2022-2025.
[6] Sugden MC,Holness MJ.Role of nuclear receptors in the modulation of insulin secretion in lipid-induced insulin resistance[J].Biochem Soc Trans,2008,36(5):891-900.
[7] Lalloyer F,Fievet C,Lestavel S,et al.The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia[J].Arterioscler Thromb Vasc Biol,2006,26(12):2731-2737.
[8] Wang Y,Lindstedt KA,Kovanen PT.Mast cell granule remnants carry LDL into smooth muscle cells of synthetic phenotype and induce their conversion into foam cells[J].Arterioscler Thromb Vasc Biol,1995,15(6):801-810.
[9] Egashira K,Inoue S,Ni W,et al.Anti-monocyte chemoattractant protein-1 gene therapy limits progression and destabilization of established atherosclerosis in apolipoprotein E-knockout mice[J].Circulation,2002,106(21):2700-2706.
[10] Sakuma H,Yamamoto M,Okumura M,et al.High glucose inhibits apoptosis in human coronary artery smooth muscle cells by increasing bcl-xL and bfl-1/A1[J].Am J Physiol Cell Physiol,2002,283(2):C422-C428.
[11] Kim JH,Jin YR,Park BS,et al.Luteolin prevents PDGF-BB-induced proliferation of vascular smooth muscle cells by inhibition of PDGF beta-receptor phosphorylation[J].Biochem Pharmacol,2005,69(12):1715-1721.
[12] Plutzky J.The PPAR-RXR transcriptional complex in the vasculature:energy in the balance[J].Circ Res,2011,108(8):1002-1016.
[13] Staels B.Regulation of lipid and lipoprotein metabolism by retinoids[J].J Am Acad Dermatol,2001,45(5):S158-S167.
[14]Streb JW,Miano JM.Retinoids:pleiotropic agents of therapy for vascular diseases?[J].Curr Drug Targets Cardiovasc Haematol Disord,2003,3(1):31-57.
[15] Chai D,Wang B,Shen L,et al.RXR agonists inhibit high-glucose-induced oxidative stress by repressing PKC activity in human endothelial cells[J].Free Radic Biol Med,2008,44(7):1334-1347.
[16] Steinberg SF.Cardiac actions of protein kinase C isoforms[J].Physiology(Bethesda),2012,27(3):130-139.
[17] Nakagawa Y.Artificial analogs of naturally occurring tumor promoters as biochemical tools and therapeutic leads[J].Biosci Biotechnol Biochem,2012,76(7):1262-1274.
[18]贾 薇,袁中华.动脉粥样硬化中的4个PKC相关酶[J].中国病理生理杂志,2007,23(7):1442-1445.
[19] Carter CA,Parham GP,Chambers T.Cytoskeletal reorganization induced by retinoic acid treatment of human endometrial adenocarcinoma(RL95-2)cells is correlated with alterations in protein kinase C-α[J].Pathobiology,1998,66(6):284-292.
[20] Radominska-Pandya A,Chen G,Czernik PJ,et al.Direct interaction of all-trans-retinoic acid with protein kinase C(PKC):implications for PKC signaling and cancer therapy[J].J Biol Chem,2000,275(29):22324-22330.
