三氟丙基修饰的二氧化硅膜制备、氢气分离及其水热稳定性能
2013-09-15洪志发李国华王学伟聂祚仁李群艳
洪志发 韦 奇*, 李国华 王学伟 聂祚仁 李群艳
(1北京工业大学材料科学与工程学院,北京 100124)
(2浙江工业大学化学工程与材料学院,杭州 310014)
微孔二氧化硅膜在不同的温度下呈现良好的氢气分离和渗透性能,在工业中有广泛的潜在应用前景[1-4],尤其是其在膜反应器中的应用,能够打破反应平衡的限制,提高产率,降低能耗,引起了研究人员的广泛关注[5-7]。然而,二氧化硅膜的水热稳定性能差,长期暴露于水热环境中其表面羟基极易吸附环境中的水汽而继续发生缩合反应并导致孔结构的崩溃,影响气体的分离效果[8-10],从而限制了其在水热环境下的长期使用。如何提高微孔SiO2膜的水热稳定性成为众多膜科学工作者的研究重点之一。目前提高膜材料水热稳定性的方法有:(1)过渡金属掺杂。Boffa等[11]研究了铌掺杂SiO2膜的水热稳定性,在200℃,水蒸气压力为56 kPa的条件下处理70 h,铌掺杂SiO2膜的H2渗透率下降了32%,而纯SiO2膜的H2渗透率下降了72%,说明铌掺杂SiO2膜比纯SiO2膜具有更稳定的骨架结构。闫建平等[12]采用正硅酸乙酯和Co(NO3)2·6H2O通过溶胶-凝胶法制备钴掺杂SiO2膜,研究表明钴元素以Si-O-Co的形式存在于SiO2骨架之中,300℃时膜材料的 H2渗透率达到 6.41×10-7mol·m-2·s-1·Pa-1。(2)通过化学修饰,即采用含有疏水基团的硅烷前驱体与硅源前驱体发生水解缩聚反应,在SiO2表面引入疏水基团,降低表面羟基浓度,提高膜材料的疏水性,降低膜材料对水分的物理吸附,从而在一定程度上避免孔结构的崩溃。1999年De Vos等[13]采用溶胶-凝胶法首次成功将甲基修饰到SiO2膜中,使SiO2膜的疏水性显著提高。李振杰等分别制备了苯基[14]和乙烯基[15,16]表面修饰的疏水SiO2膜,修饰后膜材料疏水性得到明显改善,并且具有较好的氢气渗透和分离性能。韦奇[17]等研究了甲基修饰的二氧化硅膜在长期水热环境下的H2和CO2的双组份渗透,将修饰后的二氧化硅膜在水热环境下陈化近110 h后H2渗透率基本保持不变,H2/CO2分离系数有所增大,证明经过甲基修饰的二氧化硅膜比纯二氧化硅膜具有更好的水热稳定性。
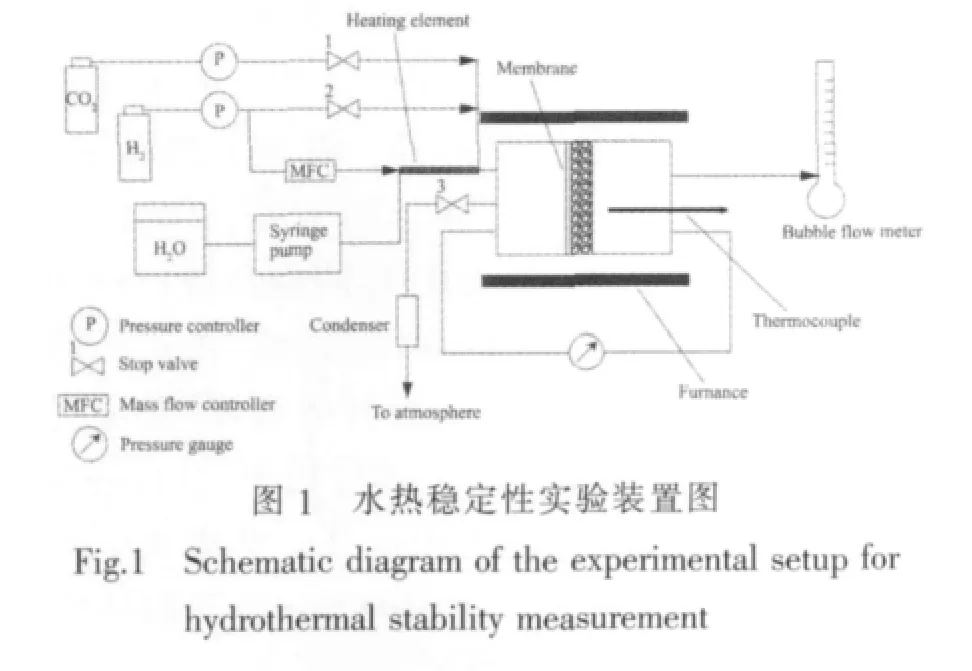
在前人的工作中,水热环境通常是采用“鼓泡”方法营造的,即让气体流过温度可控的蒸馏水,气体出来后携带一定比例的水气,水蒸气的量受水的饱和蒸汽压限制。这种环境与水煤气变换反应和甲烷水汽重整反应等化工过程的环境差别很大,不能准确地反映膜材料在上述化工过程中的水热稳定性。本文模拟水煤气变换反应环境,采用微量注射泵控制进水量,通过加热气体管道使得液态水气化,并用氢气作为载气,将水蒸气带入膜材料渗透装置,使得膜材料所处的环境更接近水煤气变换反应的水热环境。本文选择三氟丙基三甲氧基硅烷(TFPTMS)为修饰剂,通过正硅酸乙酯 (TEOS)和TFPTMS的共水解缩聚反应,制备了三氟丙基修饰的二氧化硅膜材料,并系统研究了膜材料的疏水性、孔结构、氢气渗透和分离性能以及模拟水煤气变换反应的水热环境下的稳定性。
1 实验部分
1.1 化学试剂
正硅酸乙酯(TEOS),分析纯,广东汕头市西陇化工厂;三氟丙基三甲氧基硅烷(TFPTMS),分析纯,Alfa Aesar(天津)化学有限公司;硝酸,优级纯,广东汕头市西陇化工厂;无水乙醇(EtOH),分析纯,北京化工厂;自制蒸馏水。
1.2 SiO2溶胶及膜材料的制备
采用溶胶-凝胶法制备SiO2膜,TEOS和TFPTMS作为前驱体,首先将TEOS和无水乙醇在冰水浴中进行搅拌,使其混合均匀,然后逐滴加入事先按比例混合好的硝酸和蒸馏水,继续搅拌10 min后,将其放入60℃水浴中在回流条件下搅拌反应 2.5 h, 按 nTFPTMS∶nEtOH=(0.2~0.6)∶3.8 物质的量比将三氟丙基三甲氧基硅烷与无水乙醇充分混合,逐滴加入上述溶液继续反应0.5 h,得到最终混合物的物0.085(其中,x=0,0.2,0.4 和 0.6),反应结束后得到无色透明的三氟丙基修饰的SiO2溶胶。将上述制备的溶胶和无水乙醇以1∶19体积比稀释,然后通过浸渍提拉法(Dip-coating)将其涂覆在 γ-Al2O3/α-Al2O3多孔陶瓷支撑体上。为避免空气中微颗粒附着在膜材料表面形成缺陷,整个涂膜过程在100级洁净室中进行。涂膜后在400℃下煅烧3 h,升温速率为1℃·min-1,煅烧过程中以N2作为保护气,防止氧化。为了减少和消除SiO2膜的微缺陷,上述涂膜和煅烧过程重复四次。将适量的SiO2溶胶倒入蒸发皿中干燥成凝胶,然后在400℃的N2气氛下煅烧3 h,得到无支撑三氟丙基修饰的SiO2膜。修饰后的样品标记为(x TFPTMS)SiO2(其中,x=0.2,0.4 和 0.6)。
1.3 SiO2膜的表征
SiO2膜的SEM形貌由日本日立公司S-3400N型高分辨扫描电子显微镜表征。利用Micromeritics ASPS 2020型比表面和孔隙度测量仪表征无支撑膜的孔结构,测量前将样品在300℃脱气5 h,在77.6 K测定样品的N2吸附等温线,利用Horvath-Kawazoe(H-K)模型计算并绘制样品的孔径分布曲线。采用德国Dataphysics公司OCA20视频光学接触角测量仪对膜材料进行接触角测试,室温下将体积为2μL的水滴,以1μL·s-1的注射速度滴在有支撑SiO2膜表面,然后测量水滴在膜材料表面的接触角。利用美国热电公司Nicolet 5700型傅里叶变换红外光谱仪测定纯SiO2和(0.6TFPTMS)SiO2样品的红外光谱图。固体29Si MAS NMR测试在德国Bruker AV300核磁共振谱仪上进行,测试条件为:共振频率59.62 MHz,脉冲宽度6.0μs,魔角旋转5 kHz,π/2脉冲,扫描次数200次,化学位移参照四甲基硅烷。
氢气的渗透和分离性能测定在自制的气体渗透和分离装置上完成,有支撑SiO2膜采用柔性石墨垫片密封,膜材料两端的压力差用陕西创威科技公司的精密数字压力表(CWY100)测量,通过稳压阀控制进气压力使膜材料两侧压差稳定在100 kPa,渗透端气体流量用皂泡流量计测量。H2/CO2双组份实验用气体质量流量计(MFC)来控制进气端气体的流量,用气相色谱仪(日本岛津GC2014)来测定渗透端混合气的成分及其浓度,气体的分离系数通过下列公式计算,其中Cpermeate为渗透端气体浓度,Cfeed为进样端气体浓度。

水热稳定性在自制装置上(图1)测定,营造水热环境时关闭1和2开关阀,氢气流量由气体质量流量计控制,流量设为47.5 mL·min-1,水的流量由微量注射泵控制,通过加热带后成为水蒸气,由氢气带入渗透装置内,进水量占总进气量的物质的量比例为5%,膜材料的温度维持在200℃。陈化一定时间后,通过死端渗透模型[18]分别测定膜材料的H2和CO2单组份渗透率,重复以上过程,测定膜材料经过水热条件处理不同时间后的单组份渗透率,通过研究单组份气体渗透率及理想分离系数的变化情况来表征膜材料的水热稳定性。
2 结果与讨论
2.1 有支撑体SiO2膜SEM形貌
图2为有支撑微孔SiO2膜的断面和表面SEM照片。从图2a中可以明显看出,膜体系主要由三层组成,顶层为三氟丙基修饰的SiO2膜,厚约300 nm,中间为γ-Al2O3过渡层,底层为α-Al2O3多孔陶瓷。这说明三氟丙基修饰SiO2膜已成功地浸涂到γ-Al2O3/α-Al2O3衬体上。由图2b中可以看出修饰后膜材料的表面平整光洁,没有明显的裂纹、大孔或其他严重的微缺陷。

2.2 膜材料的孔结构分析
图3为修饰前后无支撑SiO2膜的N2吸附等温线,从图中可以看出所有样品的N2吸附等温线的形状基本相同,在相对压力(p/p0<0.01)非常低时,样品的N2吸附量随着相对压力的增大而迅速升高,此时N2分子以单层吸附的方式吸附于孔表面,表明修饰前后膜材料的孔结构较为发达,而且微孔含量很高。当 0.01<p/p0<0.2 时,N2吸附量缓慢上升,当p/p0>0.2吸附等温线趋于水平,吸附达到饱和。吸附等温线属于IUPAC分类的第一类等温线,说明修饰前后样品的孔结构均为典型的微孔结构。



Pore volume/(cm3·g-1)SiO2 444.24 0.22(0.2TFPTMS)SiO2 347.16 0.17(0.4TFPTMS)SiO2 309.04 0.15(0.6TFPTMS)SiO2 273.36 0.14 Sample BET surface area/(m2·g-1)
图4是修饰前后样品的孔径分布图,如图所示,修饰前后样品的孔径分布范围基本一致,孔径集中分布在0.45~0.7 nm之间,说明三氟丙基的修饰并没有对孔径产生很大的影响。从表1可以看出随着三氟丙基修饰量的增加,样品的孔容和比表面积逐渐减少,其原因可能是由于三氟丙基接枝在孔道表面,占据了部分空间,导致孔容和比表面积减少,相应的N2吸附量也会减少,这与N2吸附等温线的结果是一致的。对比苯基[14]和乙烯基[15]修饰的微孔SiO2膜材料,修饰后的样品的孔容和比表面都出现不同程度的减少,说明修饰基团对膜材料孔容和比表面有一定的影响。
2.3 SiO2膜的疏水性能
图5是修饰前后有支撑SiO2膜对水的接触角测量结果。从图中可以看出,水滴在未修饰SiO2膜表面的接触角只有(33.8°±1.0°)(图 5a),随着 TFPTMS加入量的增加, 接触角不断增大 (图5b、5c、5d),当nTFPTMS/nTEOS=0.6 时,膜材料对水的接触角达(102.7°±0.1°)。膜材料由亲水变成疏水,这是由于膜表面的部分羟基被疏水的三氟丙基所取代,膜材料的疏水性能得到显著提高。

为了进一步说明三氟丙基的成功修饰,我们对比了修饰前后SiO2膜的FT-IR谱图。图6为修饰前后的SiO2膜的FT-IR谱图。从图中可以看到波数799 cm-1和1 074 cm-1处的吸收峰分别为Si-O-Si对称和反对称伸缩振动特征峰,1 630 cm-1和3 442 cm-1处分别对应于表面羟基和物理吸附水的吸收峰。对于三氟丙基修饰的样品,波数1 221 cm-1和1 268 cm-1处对应于-CF3伸缩振动峰,1 451 cm-1、1 379 cm-1、1 321 cm-1处为-CH2-的振动特征峰,903 cm-1和840 cm-1处对应于-SiC-振动峰,而未修饰的SiO2样品在上述波数处均未出现吸收峰,这说明了三氟丙基已成功修饰到SiO2膜材料中,且经过400℃的煅烧,碳氟基团并没有被破坏。从图6中还可以看到未修饰的SiO2膜在3 442 cm-1处的物理吸附水的吸收峰非常强烈,而三氟丙基修饰后的膜材料吸附水的吸收峰较弱,说明经修饰后膜材料的疏水性得到增强。这与图5接触角实验结果一致。同时可以看出三氟丙基修饰后的膜材料中Si-OH在1 630 cm-1处的吸收峰明显减弱,表明Si-OH浓度有明显的降低,说明三氟丙基的存在取代了部分羟基,使膜材料表面由亲水性变为疏水性。

为了定量描述修饰前后膜表面的化学性质,我们对无修饰的SiO2膜和(0.6TFPTMS)SiO2膜材料进行了固体29Si MASNMR分析,并对图谱进行高斯拟合分峰处理,结果如图7所示。从纯SiO2膜的NMR图上看出,在-92、-102、-110 ppm处有3个共振峰,分别代表的是 Q2-Si(i.e.Si(OSi≡)2(OH)2)、Q3-Si(i.e.Si(OSi≡)3(OH))、Q4-Si(i.e.Si(OSi≡)4)的共振峰[19-20],修饰后样品的NMR图在-102、-110 ppm处出现Q3、Q4共振峰,但没有出现Q2共振峰,在-60和-67 ppm处多了2个共振峰,分别代表T2-Si(i.e.Si(OSi)2(OH)R R=-CH2CH2CF3)以及 T3-Si(i.e.Si(OSi)3R)的信号[19-22]。根据Q峰和T峰的峰强度可以计算出修饰前后SiO2膜材料中羟基的浓度,以及修饰后SiO2膜材料中三氟丙基的浓度,单位mmol·g-1,结果如表2所示。从表中数据可以看出修饰后的SiO2膜三氟丙基的浓度为3.42 mmol·g-1,且其表面羟基的浓度从 10.3 mmol·g-1下降到 5.06 mmol·g-1。 结合接触角和红外实验的结果不难证明,三氟丙基的存在和羟基浓度的降低是修饰后SiO2膜材料的疏水性能得到提高的原因。而且Q2峰的缺失证明了三氟丙基的存在使表面羟基的浓度下降到最低[23]。

2.4 气体渗透和分离性能
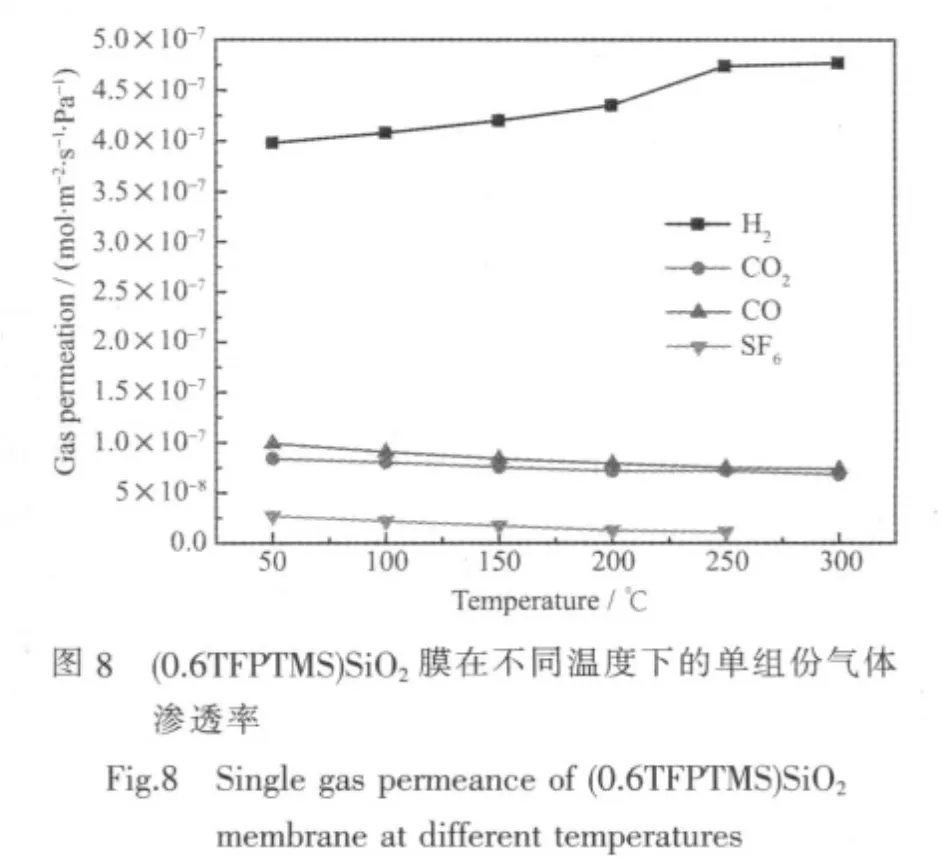

表2 修饰前后SiO2膜的固体29Si MASNMR的实验结果Table 2 Solid state 29Si MASNMR results of the pure and modified silica membranes

表3 (0.6TFPTMS)SiO2膜的单组份理想分离系数和双组份分离系数Table 3 Permselectivity and separation factor of(0.6TFPTMS)SiO2 membrane
图8为有支撑三氟丙基修饰SiO2膜在不同温度下H2、CO2、CO和SF6的单组份渗透率。表3为不同 温 度 下 H2/CO、H2/CO2、H2/SF6、CO/SF6和 CO2/SF6的理想分离系数及进气量为50/50(mL·min-1)的H2/CO2双组份混合气体分离系数,并且与各自的Knudsen扩散分离因子作比较。从图8可以看出,H2的渗透率随着温度的升高逐渐增大,300℃时达到4.77×10-7mol·m-2·s-1·Pa-1,从表 3 可以看出 H2/CO2、H2/CO、H2/SF6的理想分离系数随温度的升高而升高并且均高于各自的Knudsen扩散分离因子,说明H2在膜材料中的输运行为突破了Knudsen扩散的限制,为典型的微孔扩散机理。膜材料在不同温度下对H2/CO2、H2/CO和H2/SF6的分离起到了分子筛分作用。SF6的分子动力学直径为0.55 nm,一般认为SF6只能在尺寸大于0.55 nm的微孔或介孔甚至微缺陷中输运,从图中可以看出SF6的渗透率随着温度的升高而降低,说明SF6的输运受到Knudsen扩散机制的控制。而H2,CO2和CO气体分子既能进入到小于0.55 nm的微孔中输运,也能在大于0.55 nm的微孔或介孔甚至微缺陷中输运,从图7和表3可以看出CO2和CO的单组份渗透率随温度的升高而降低,而不同温度时CO/SF6和CO2/SF6的分离系数均大于各自的Knudsen扩散分离因子,说明CO和CO2气体的输运既遵循微孔扩散机理,也受到Knudsen扩散机制的控制。
从表3可以看出H2/CO2双组份混合气体分离系数在不同温度下均高于Knudsen扩散分离因子,且与单组份理想分离系数的规律基本一致,说明在实际混合气体中H2和CO2的渗透分离同样遵循微孔扩散机理,而在100℃到250℃之间H2/CO2双组份气体分离系数略高于单组份气体理想分离系数,可能的原因是H2和CO2在孔道内输运存在竞争机制,使分离系数有所提高。
2.5 水热稳定性研究
图9表示修饰后膜材料经过200℃,水蒸气含量为5%的水热环境处理不同时间后的H2和CO2单组份渗透率及H2/CO2理想分离系数,从图中可以看出前 3 h,H2的渗透率从 5.5×10-7mol·m-2·s-1·Pa-1下降到 5.0×10-7mol·m-2·s-1·Pa-1, 降幅较小,仅为9.1%, 然后基本稳定在 4.9×10-7mol·m-2·s-1·Pa-1左右,CO2的渗透率基本保持不变,起始H2渗透率的下降可能是因为样品接触水蒸气后吸附了一定量的物理吸附水,吸附水占据了一定的空间[24],吸附平衡之后孔结构不再改变,所以通过微孔扩散的H2和CO2的渗透率也就基本保持不变。H2/CO2理想分离系数先升后降并且最后基本稳定在4.9左右,均超过H2/CO2Knudsen扩散分离因子4.69。而分离系数一开始的波动可能是因为样品接触到水蒸气孔结构发生微小的调节,微调结束之后孔结构不再改变,H2和CO2的渗透率基本保持不变,理想分离系数也就达到平衡。前人的研究表明,在水热环境下,纯SiO2膜的H2渗透率和H2/CO2分离系数都随着陈化时间的推移而急剧下降[25],与纯SiO2膜相比,经过三氟丙基修饰的SiO2膜表现出更优异的水热稳定性。

3 结 论
采用溶胶-凝胶法,通过三氟丙基三甲氧基硅烷(TPFTMS)和正硅酸乙酯(TEOS)的共水解缩聚反应成功制备了三氟丙基修饰的SiO2膜,三氟丙基的修饰提高了膜材料的疏水性,当nTFPTMS/nTEOS=0.6,膜材料对水的接触角达到(102.7°±0.1°)。膜材料保持良好的微孔结构,孔径狭窄分布在0.45~0.7 nm之间。氢气在有支撑三氟丙基修饰的SiO2膜的输运遵循微孔扩散机制,300℃时H2的单组份渗透率达到4.77×10-7mol·m-2·s-1·Pa-1,H2/CO2的理想分离系数为6.99,双组份混合气体分离系数为6.93。膜材料在200℃,水蒸气物质的量含量为5%的水热环境中具有良好的稳定性,能够连续工作220 h以上。
[1]De Vos R M,Verweij H.Science,1998,279:1710-1711
[2]Asaeda M,Yamasaki S.Sep.Purif.Technol.,2001,25:151-159[3]Anita R,Sunil A,Yang SS.Renew.Energ.,2010,35:2649-2655
[4]Kanezashi M,Shioda T,Gunji T,et al.AIChE J.,2012,58:1733-1743
[5]HUANG Zhong-Tao(黄仲涛),ZENG Zhao-Huai(曾昭槐),ZHONGBang-Ke(钟邦克),et al.Technology and Application of Inorganic Membrane(无机膜技术及其应用).Beijing:China Petrochemical Press,2002:455-456
[6]Giessler S,Jordan L,da Costa D J C,et al.Sep.Purif.Technol.,2003,32:255-264
[7]WANG Wei-Ping(王卫平),PAN Xiu-Lian(潘秀莲),ZHANG Xiao-Liang(张小亮),et al.Chinese J.Catal.(Cuihua Xuebao),2005,26(12):1042-1046
[8]Castricum H L,Sah A,Kreiter R,et al.Chem.Commun.,2008,9:1103-1105
[9]WEI Qi(韦奇),LI Jian-Lin(李建林),SONG Chun-Lin(宋春林),et al.J.Inorg.Mater.(Wuji Cailiao Xuebao),2004,19(1):133-139
[10]QI Hong(漆虹),HAN Jing(韩静),JIANG Xiao-Luo(江晓骆),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,25(7):758-764
[11]Boffa V,Blank D H A,ten Elshof J E.J.Membr.Sci.,2008,319(1-2):256-263
[12]YAN Jian-Ping(闫建平),WEI Qi(韦奇),DUAN Xiao-Yong(段 小 勇 ),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(7):1334-1340
[13]De Vos R M,Maier W F,Verweij H.J.Membr.Sci.,1999,158:277-278
[14]LI Zhen-Jie(李振杰),WEI Qi(韦奇),WEI Na-Na(魏娜娜),et al.Chem.J.Chinese Universities(Gaodeng Xuexiao Huaxue Xuebao),2010,31(12):2482-2487
[15]WANG Yan-Li(王艳丽),WEI Qi(韦奇),YU Chun-Xiao(于春晓),et al.J.Inorg.Mater.(Wuji Cailiao Xuebao),2007,22(5):949-953
[16]Wei Q,Wang Y L,Nie ZR,et al.Micropor.Mesopor.Mater.,2008,111:97-103
[17]WEI Qi(韦奇),LI Jian-Lin(李建林),SONG Chun-Lin(宋春林),et al.J.Inorg.Mater.(Wuji Cailiao Xuebao),2004,19(2):417-423
[18]Li G,Kanezashi M,Tsuru T.J.Membr.Sci.,2011,379:287-295
[19]Chong A S M,Zhao X S,Kustedjo A T,et al.Micropor.Mesopor.Mater.,2004,74:33-42
[20]Wang Y Q,Yang C M,Zibrowius B,et al.Chem.Mater.,2003,15:5029-5035
[21]Wei Q,Wang F,Nie Z R,et al.J.Phys.Chem.B,2008,112:9354-9359
[22]Wei Q,Chen H Q,Nie Z R,et al.Mater.Lett.,2007,61:1469-1473
[23]Yang D J,Li J P,Xu Y,et al.Micropor.Mesopor.Mater.,2006,95:180-186
[24]Saito T,Seshimo M,Akamatsu K,et al.J.Membr.Sci.,2012,329:95-100
[25]Dong J H,Lin Y S,Kanezashi M,et al.J.Appl.Phys.,2008,104:121301
