肉桂酸及其衍生物的Euバ配合物的合成、结构及荧光性质研究
2013-09-15周贤菊周凯宁王志亮肖腾蛟赵小奇李清旭
周贤菊 周凯宁 王志亮 肖腾蛟 赵小奇 李清旭
(1重庆邮电大学数理学院,重庆 400065)
(2东华大学材料科学与工程学院,上海 201600)
0 引 言
金属-有机配合物具有特殊的结构,拥有优异的光、电、磁学性能,并在催化、分离、吸附、气体存储和发光材料方面有广阔的应用前景[1-5],因而备受关注,是当今研究的一个热点课题。尤其是近年来生物荧光探针和电致发光金属有机配合物的研发[6-8],更推动了该领域的发展。金属-有机配合物所用配体涉及到β-二酮[9]、羧酸及其衍生物[10]、多环有机物[11]、穴状物[12]、蛋白质[13]等,其中苯甲酸类有机配体因配位能力强、体系共轭程度大、来源方便等原因被作为多种过渡金属和稀土离子的有机配体,得到广泛的研究[14-18]。阎冰等[15]研究了稀土离子与苯甲酸及其衍生物的合成、发光性能和分子间的能量传递,Oyang等[17]研究了对位氨基苯甲酸铽的晶体结构、光学性质和第二配体对发光性能的影响。但配体取代基对配合物晶体结构、堆积方式和物化性能影响的研究,还比较少。肉桂酸比苯甲酸多出一个环外双键,是一种很好的半刚性有机配体。肉桂酸(HL)具有广泛的抗实体肿瘤活性,能抑制肿瘤的增殖并诱发其分化,因此肉桂酸-稀土配合物具有重要的研究意义和潜在应用前景。杨艳红等采用水热法合成了Eu、Tb、Sm和Gd与肉桂酸的配合物[19]。过渡金属Cu和Cd与肉桂酸的配位也有报道[20]。本文选用了肉桂酸及对位取代衍生物R-L(R=CH3Cl,NO2,OCH3,OH)为配体,制备了 6 种不同配体的 Eu肉桂酸配合物,研究了其晶体结构、光学性能及取代基性质对配合物的影响。
1 实验部分
1.1 试剂与仪器
试剂:Eu2O3(99.99%),肉桂酸及其衍生物为分析纯,其他试剂包括盐酸、稀氨水、DMF(N,N-二甲基甲酰胺)和去离子水。红外吸收光谱采用KBr压片技术在美国PerkinElmer仪器有限公司生产的Spectrum 65傅里叶变换红外光谱仪上记录;单晶结构测定在牛津Oxford Gemini SUltra型X-射线单晶衍射仪上完成;紫外吸收光谱使用北京普析通用仪器设备公司TU-1901型紫外可见分光光度计;荧光光谱利用英国Edinburgh FLS920稳态瞬态荧光光谱仪测得,其光源为450 W氙灯光源,探测器为Shimidazu R9287 PMT。所有测试均在室温下进行。

表1 系列配合物的红外光谱主要特征吸收峰Table 1 Main characteristic absorption bands of FT-IR spectra for the series of complexes
1.2 样品合成
配合物对甲基肉桂酸铕(1),对氯肉桂酸铕(2),对硝基肉桂酸铕(3),对甲氧基肉桂酸铕(4),对羟基肉桂酸铕(5),肉桂酸铕(6)的制备方法类似:称取1.5 mmol Eu2O3,溶解于5 mL 37%盐酸中,蒸发至干。用10 mL的水溶解残留物,通过添加稀氨水调节pH值至8。随后将沉淀过滤,水洗,晾干。将沉淀加入到溶解有10 mmol肉桂酸或肉桂酸衍生物的DMF溶液中搅拌8 h后过滤。将滤液装入烧杯,蜡封,在室温下置于黑暗的通风橱内结晶。本研究得到无色针状晶体1,无色片状晶体2和淡黄色块状晶体3。
1.3 晶体结构的测定
使用牛津Oxford Gemini SUltra型X射线单晶衍射仪,用经石墨单色仪单色化的Mo Kα射线(λ=0.071 073 nm)。3种配合物相关衍射数据分别为:配合物 1:衍射总数:24 794;独立衍射数:13 786,Rint=0.029;可观测衍射数:10 326(I>2σ(I))。配合物 2:衍射总数:22 015; 独立衍射数:12 555,Rint=0.035;可观测衍射数:9 651(I>2σ(I))。配合物 3:衍射总数:17 668;独立衍射数:7 559,Rint=0.047;可观测衍射数:6 376(I>2σ(I))。配合物采用相同的测试方法和数据处理方式。所得数据经Lp因子及经验吸收校正。晶体结构采用直接法解出。对氢原子和非氢原子分别采用各向同性和各向异性温度因子进行全矩阵最小二乘法修正。配合物的结构解析使用SHELXL-97程序完成。
CCDC:856461,1;856460,2;856462,3。
2 结果与讨论
2.1 配合物的红外吸收光谱分析
配合物及对应配体的红外光谱在常温下用KBr压片,扫描范围为450~4 000 cm-1,扫描次数为30次。配合物的主要特征吸收峰如表1所示。
将配合物红外吸收光谱与相应配体红外吸收光谱比较后发现:在配合物中,与羧酸相关的羧酸二聚体伸缩振动峰这是由于配合物的羧酸根离子与Eu3+配位后,由于对称和非对称伸缩振动所产生的。由此可以说明配体的羧酸根离子参与了配位[23]。另外,在配合物1~5中发现,在1 660 cm-1左右出现了1个强而锐的羰基振动(νC=O)吸收峰,相对于游离DMF的羰基伸缩振动(νC=O=1 670 cm-1)吸收向低波数移动;同时,在1 400 cm-1附近处出现了νC-N伸缩振动峰,表明这5个配合物中存在配位DMF分子,通过羰基氧原子与Eu3+配位。而配合物6中未发现这2个峰,推测DMF未参与配位。另外,还可以从配合物在3 000~3 700 cm-1范围宽峰的形状和强度推测水分子配位情况,所得结果可与单晶X射线衍射分析相互应证。
2.2 配合物单晶结构描述
本研究获得3种配合物单晶,配合物1和2结构类似,均为一维长链。配合物3为双核结构,这里分别对3种配合物结构进行描述。
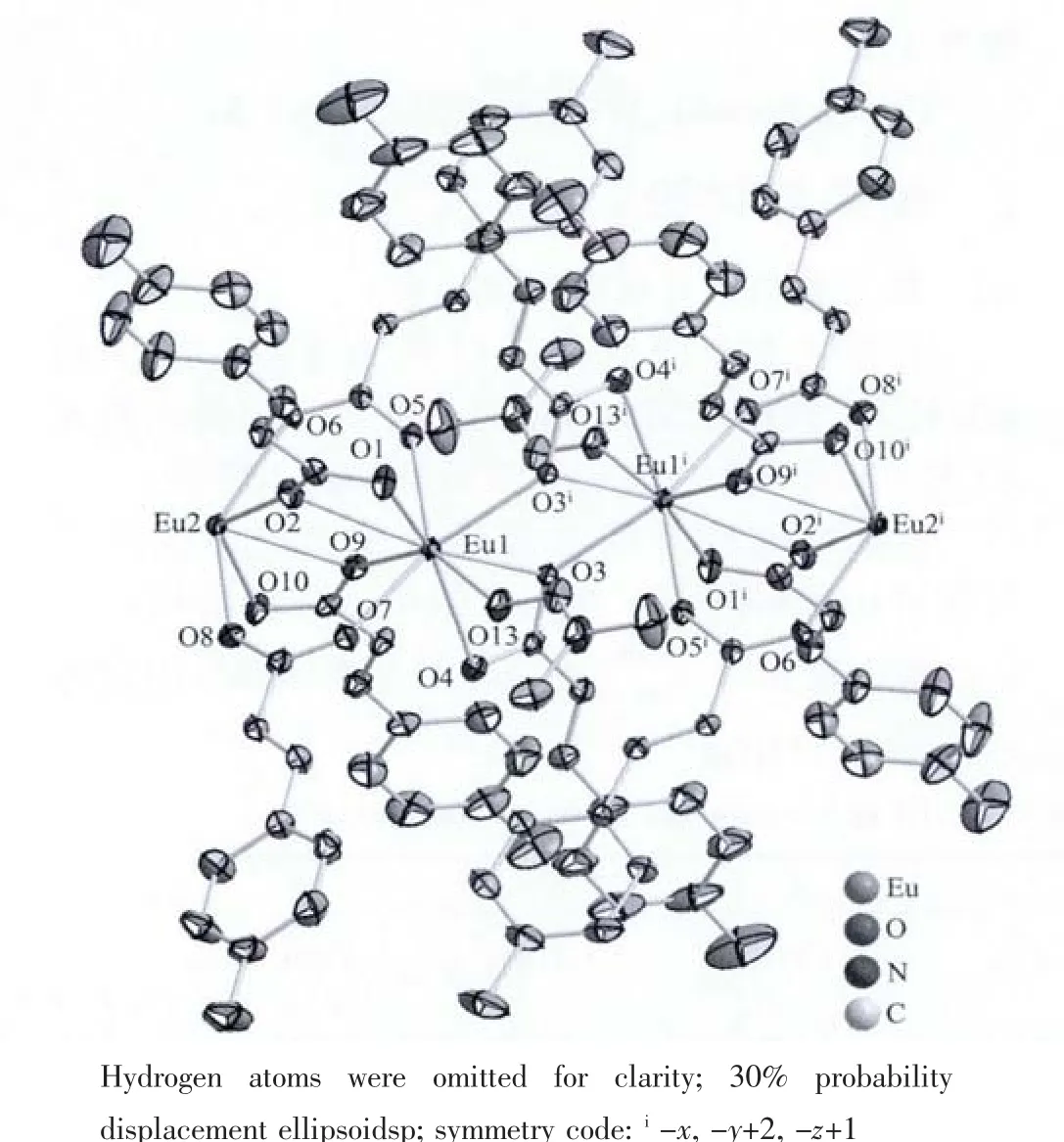
图1 配合物1的分子结构单元图Fig.1 Structure of complex 1
配合物1为无限长单链结构,其分子式可以写成{[Eu2(CH3-L)6(DMF)(H2O)]·2DMF·H2O}n。 其分子结构单元如图1所示。配合物中存在2种具有不同配位形式的Eu3+离子(Eu1和Eu2)。每1个Eu1与9个氧原子配位,其中6个氧原子来自4个通过三齿螯合桥接方式配位的配体羧酸根离子;2个氧原子来自2个通过双齿桥接方式配位的配体羧酸根离子;另外1个氧原子来自单齿配位的DMF。每1个Eu2同样与9个氧原子配位。其中,6个氧原子来自4个通过三齿螯合桥接方式配位的配体羧酸根离子;2个氧原子来自2个通过双齿桥接方式配位的配体羧酸根离子;另外1个氧原子来自单齿配位的水分子。Eu1周围9个氧原子构成1个扭曲的单帽四方反棱柱,O2构成帽顶,O5、O9、O1和O7构成其上表面,O13、O3、O4和 O3i组成其下表面。 Eu2周围 9个氧原子也构成1个扭曲的单帽四方反棱柱,和Eu1构成的多面体的区别在于扭曲程度和方向不同(图 2)。 O10 构成帽顶,O8、O9、O11A、和 O14 组成其上表面,O2、O6、O11和O12组成其下表面。Eu3+与羧酸基团的氧原子形成的Eu-O键的键长范围在 0.234 1~0.266 1 nm 之间,Eu-O(H2O)为 0.242 1(3)nm,Eu-O(DMF)为 0.238 9(3)nm。Eu1 和 Eu1i(Eu2 和Eu2i)之间通过2个配体三齿螯合桥接;Eu1和Eu2之间通过4个配体桥接,其中2个为三齿螯合桥接,另外2个为双齿桥接。这样就形成了…Eu1iEu1Eu2Eu2i…型的无限长单链状配合物(图3)[24-25]。在配合物(1)中同时观察到了4个氢键,其相关数据见表2。在这些氢键中,O17-H17A…O15和O17-H17B…O15,给体都是结合水分子,受体为结合的DMF分子中的氧原子,通过这2个氢键使水分子和DMF分子与配合物结合。
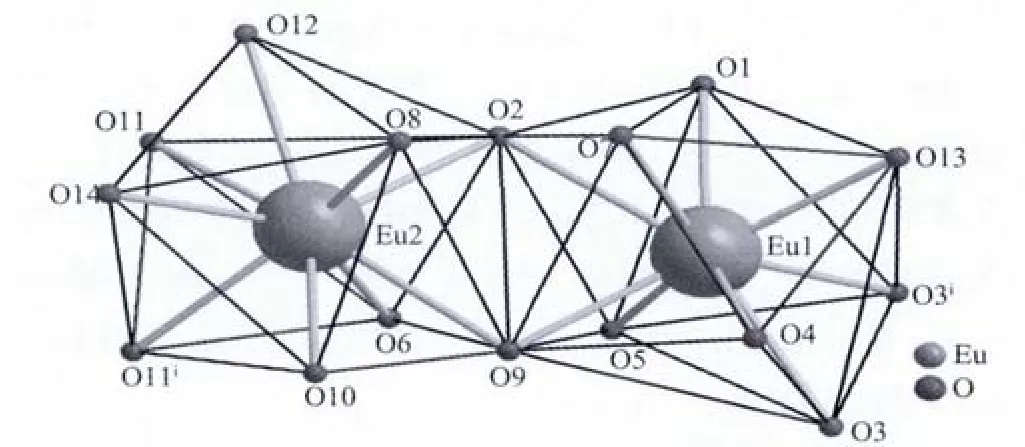
图2 配合物1中心金属离子配位多面体构型Fig.2 Coordination polyhedrons of Eu1 and Eu2 in complex 1

图3 配合物1的一维链状结构Fig.3 1D chain of complex 1

表2 配合物的氢键键长和键角Table 2 Hydrogen bonds and bond angles for complexes
配合物 2({[Eu2(Cl-L)6(DMF)(H2O)]·2DMF·H2O}n)的椭球结构图如图4所示。通过X-射线衍射实验结果发现配合物2与配合物1的结构类似,均为无限长单链结构,配合物中也存在2种具有不同配位形式的铕离子Eu3+(Eu1和Eu2),每1个Eu3+均与 9个氧原子配位,只是配位形式与配合物1有少许差别。由于篇幅限制,我们在这里不再赘述配合物2的具体结构。


配合物3为双核结构,其分子式可以写成Eu2(NO2-L)6(DMF)2(H2O)2·2DMF,其分子结构如图 5 所示。配合物中每1个Eu3+和9个氧原子配位,其中与4个来自2个对硝基肉桂酸羧酸根离子的氧原子通过双齿螯合方式配位;与3个来自2个对硝基肉桂酸羧酸根离子的氧原子通过三齿螯合桥接方式配位,与2个分别来自DMF和水分子的氧原子单齿配位。Eu3+周围9个氧原子构成1个扭曲的单帽四方反棱柱,O10 构成帽顶,O1、O9、O10i和 O13构成其上表面,O2、O5、O6和O14组成其下表面。Eu3+与羧酸基团的氧原子形成的Eu-O键的键长范围在 0.234 6(4)~0.277 5(5)nm 之间,Eu-O(H2O)为0.242(4)nm,Eu-O(DMF)为 0.234 6(4)nm,Eu1i和Eu1之间通过2个对硝基肉桂酸配体三齿螯合桥接形成双核结构,各分子之间通过范德华力作用形成单晶结构;在结构中同时观察到了4个氢键 (表2)。 O13-H13A…O6ii和 O13-H13B…O1i为分子内氢键,它使配合物结构更加稳定。
2.3 配合物的UV-Vis吸收光谱
分别将配合物和相应配体溶于无水乙醇中,配制成浓度为 10-5mol·L-1的溶液,在 190~400 nm 范围内扫描UV-Vis吸收光谱。以配合物2为例,吸收光谱见图6,其他配合物及其配体的吸收光谱类似配合物2,其吸收峰值列于表3。

图6 配合物2及其配体的UV-Vis吸收光谱Fig.6 UV-Vis absorption spectra of complex 2 and its corresponding ligand
从紫外吸收光谱可以得知,配体和配合物均有2个宽峰吸收带,其中,位于短波长区间的为E2吸收带,长波长区间的为B吸收带,两者都是由共轭体系的π-π*跃迁引起[26]。配体和配合物的E2吸收带没有发生较大变化,但B带却发生明显位移。除配合物2以外,其他5种配合物的B带产生5~8 nm的显著蓝移,这是由于羧酸根离子和Eu3+配位后,解除了原来的C=O和配体共轭体系的π-π共轭,降低了体系的共轭程度,从而导致吸收波长蓝移[27]。而配合物3的B带吸收相对其配体却有一定红移,这是由于羧酸根离子和Eu3+配位后,解除了原来的C=O和C=C键的π-π共轭,且由于硝基是强吸电子基,当配位以后,吸电子性被解除,体系的电偶极矩变大,使体系能量升高,容易被激发,从而导致吸收波长红移[28]。这也有力的证明了配体和稀土铕离子的配位(表3)。

表3 Eu系列配合物及其配体的UV-Vis吸收峰Table 3 UV-Vis absorption bands of the complexes and ligands
2.4 配合物的荧光光谱
配合物的荧光光谱在室温下用英国艾丁伯格FLS920型稳态/瞬态荧光光谱仪测定,激发和发射狭缝均为0.4 nm。通过监测Eu3+离子的5D0→7F2的发射,得到配合物的激发光谱。以配合物1为例,监测618 nm的发射波长,在200~550 nm范围内得到样品的激发光谱;用280~330、395、463 nm等多个波长可有效激发样品,在550~750 nm范围内得到配合物的发射光谱,如图7所示。可以看出:配合物1的激发光谱由三部分组成,第一部分为331 nm附近出现的强的吸收带,归属为Eu3+-O2-的电荷迁移带跃迁,即Eu3+与近邻的O2-形成Eu3+-O2-键合,O2-的电子从2p轨道上迁移至Eu3+的4f壳层,或称LMTC配体到金属的电荷迁移跃迁;第二部分为361、374、394和464 nm处的锐峰,分别归属为Eu3+的 f-f跃迁,7F0→5D4、7F0→5G6、7F0→5L6和7F0→5D2[29];第三部分为250~450 nm范围内的宽峰,归属为激发光对配体的激发[30]。这个范围内,配体吸收能量后,将能量传递给中心稀土离子,稀土离子通过特征发射将能量释放,这就是所谓的 “天线效应”(Antenna effect)。“天线效应”不但提高了配体与稀土离子之间的能量传递的效率,增强了稀土离子本身的发射强度,而且显著的拓宽了稀土离子对激发光的选择性,大大降低了稀土离子对激发光源的单色性的要求。

图7 配合物1的室温激发和发射光谱Fig.7 Excitation and emission spectra of complex 1 at room temperature
配合物的发射光谱中,能够观察到的发射有4组锐峰,位于579、593、618和697 nm。这4组峰分别归属于5D0→7F0、5D0→7F1、5D0→7F2和5D0→7F3。 其中,5D0→7F1为磁偶极跃迁,5D0→7F2为电偶极跃迁。5D0→7F2比5D0→7F1强很多,说明稀土离子处于非对称反演中心,即Eu3+处于无反演对称中心的配位环境中,这一点在单晶结构分析中也得到了证明。而光谱选择定则禁阻的5D0→7F0跃迁在该光谱中也被观察到,尽管非常弱,更说明Eu3+所处配位环境的对称性很低。单晶X射线分析结果表明配合物2中有2种不同格位的Eu3+离子,即Eu1和Eu2,但在该配合物的室温发射光谱中没有观察到2个5D0→7F0跃迁(579 nm处没有分裂),在低温荧光光谱中仍没有观测到分裂[14],推测可能原因是Eu1和Eu2的配位环境差异微小。另外,在室温真空情况下,分别测量了6种样品的荧光寿命。检测Eu3+在618 nm的特征激发,激发波长采用250、260或者340 nm的配体激发波段避开Eu3+的特征激发,结果拟合得到2个寿命τ1和τ2,分别归属为配体中心的荧光寿命和 Eu3+的荧光寿命。对于 1 τ1=110 μs,τ2=338 μs,对于 2 τ1=97 μs,τ2=371 μs, 对于 3 τ1=7.4 μs,τ2=18.4 μs,对于 4 τ1=119 μs,τ2=241 μs,对于 5 τ1=189 μs,τ2=545 μs,对于 6 τ1=190 μs,τ2=690 μs。
其他配合物的荧光光谱与配合物1相似,主要区别在于发光强度不同和Eu3+特征峰的位置有少许位移。在保持相同的激发狭缝和发射狭缝宽度情况下,记录到6种配合物的荧光发射强度排序由强到弱为:肉桂酸铕6,对羟基肉桂酸铕5,对氯肉桂酸铕2,对甲氧基肉桂酸铕4,对甲基肉桂酸铕1,对硝基肉桂酸铕3。造成荧光强度差异的主要原因是对位取代基的电子效应不同将不同程度改变配体的三重态能级,造成三重态和中心离子激发态匹配程度不同,最终造成不同的传递效率、荧光强度、荧光寿命差异,其进一步的研究工作正在进行中。另外,配合物6中没有配位的溶剂小分子,降低了小分子振动造成的荧光猝灭;也没有对位取代基带来的C-H,O-H的振动和荧光猝灭,所以配合物6荧光强度最强。
3 结 论
本文主要报道了6种Eu3+-肉桂酸配合物的合成、结构和光谱性能,并研究了肉桂酸苯环上不同对位取代基对配合物性能的影响。晶体结构表明,配合物形成了一维链状或双核结构。在配合物1、2、3中,Eu3+配位数位为9,且有2种不同的配位环境,形成了2个晶体学不等价格位。红外光谱证明了羧酸的解离和羧酸根离子的配位,紫外-可见吸收光谱观察到对位取代配体的B带吸收带因取代基电子效应影响较肉桂酸的B带吸收带均发生红移,配合物的B带吸收带因羧酸离子与Eu3+配位而发生明显红移(3)或蓝移(1,2,4,5,6)。 光致荧光光谱发现,配体激发峰能够有效激发配合物,说明配合物中有明显的天线效应,发生了配体到金属的能量传递(LMET)。而强吸电子性的对位取代硝基使得配体三重态能级升高,从而使三重态与中心离子激发态能级匹配程度降低,传递效率下降,造成荧光强度和荧光效率降低明显。
[1]Lammle K,Trevethan T,Schwarz A,et al.Nano Lett.,2010,10:2965-2971
[2]Hang T,Zhang W,Ye H Y,et al.Chem.Soc.Rev.,2011,40:3577-3598
[3]Llqwellyn P L,Horcajada P,Maurin G,et al.J.Am.Chem.Soc.,2009,131:13002-13008
[4]Binnemans K.Chem.Rev.,2009,109:4283-4374
[5]Bünzli J C G,Piguet C.Chem.Soc.Rev.,2005,34:1048-1077
[6]Strasser CA,Chien CH,Lopez M D G,et al.Angew.Chem.Int.Ed.,2011,50:946-950
[7]Tang H,Tang H,Zhang Z,et al.Synth.Metals,2009,159:72-77
[8]Li C Y,Zhang X B,Qiao L,et al.Anal.Chem.,2009,81:9993-10001
[9]Vigato P A,Peruzzo V,Tanburini S.Coord.Chem.Rev.,2009,253:1099-1201
[10]Singh J,Dutta PK.J.Macro.Sci.A,2011,48:246-253
[11]Gao W Y,Niu Y,Chen Y,et al.Cryst.Eng.Comm.,2012,14:6115-6117
[12]Alliger G E,Müller P,Do L H,et al.Inorg.Chem.,2011,50:4107-4115
[13]Riley K E,Hobza P.Cryst.Growth Des.,2011,11:4272-4278
[14]Lam A W H,Wong W T,Gao S,et al.Eur.J.Inorg.Chem.,2003:149-163
[15]Yan B,Zhang H J,Wang S B,et al.J.Chin.Chem.Soc.,1997,44:567-573
[16]Arnaud N,Georges J.Analyst,2000,125:1487-1490
[17]Oyang L,Sun H L,Wang X Y,et al.J.Mole.Struc.,2005,740:175-180
[18]Roh SG,Oh JB,Nah M K,et al.Bull.Korean Chem.Soc.,2004,25:1503-1507
[19]YANG Yan-Hong(杨艳红),LIYe(李野),NIU Shu-Yun(牛淑云),et al.Chin.J.Appl.Chem.(Yingyong Huaxue),2010,27:1055-1059
[20]MAOJia-Yuan(茅嘉原),FANGHong-Xin(方红新),XUQing-Feng(徐庆峰),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24:1046-1050
[21]Lu Y,Miller JD.J.Colloid Interface Sci.,2002,256:41-52
[22]Kariakin A,Davydov D,Peterson J A,et al.Biochem.,2002,41:13514-13525
[23]Cui Z,Ren H,Liu G,et al.J.Rare Earths,2006,24:724-727
[24]Seward C,Hu N X,Wang S.J.Chem.Soc.,Dalton Trans.,2001:134-137
[25]Manson JL,Arif A M,IncarvitoC C D,et al.J.Solid State Chem.,1999,145:369-378
[26]Pretsch E,Bühlmann P,Affplter C.Translated by RONG Guo-Bin(荣国斌).Structure Determination of Organic Compounds Tablesof Spectral Data(波谱数据表有机化合物的结构解析).Shanghai:Shanghai University of Polytechnology Press,2002:397
[27]Yang W,Hou Q,Liu C,et al.J.Mater.Chem.,2003,13:1351-1355
[28]Li Y,Zou Y.Adv.Mater.,2008,20:2952-2958
[29]ZHOU Xian-Ju(周贤菊),CHEN Jia(陈加),YANG Xiao-Dong(杨小东).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28:932-936
[30]Erostyák J,Buzády A,Hornyák I,et al.J.Photochem.Photobio.A,1998,115:21-26
