正反胶束体系中木素过氧化物催化氧化藜芦醇条件优化
2013-09-07方振敏袁兴中曾光明韩增辉郭灵芝黄华军湖南大学环境科学与工程学院湖南长沙410082湖南大学环境生物与控制教育部重点实验室湖南长沙410082
方振敏,袁兴中*,曾光明,韩增辉,郭灵芝,彭 馨,刘 薇,黄华军 (1.湖南大学环境科学与工程学院,湖南 长沙 410082;2.湖南大学环境生物与控制教育部重点实验室,湖南 长沙 410082)
环境中木质素类物质的大量存在使得木质素降解酶的研究具有现实意义[1].木素过氧化物酶(Lip)被认为是木质素降解过程中的关键酶类[2],它是由具有木素降解功能的白腐真菌分泌的胞外酶[3],可以催化一系列酚型和非酚型的木素模型化合物.在这些模型化合物中,非酚型富含电子的芳香族化合物藜芦醇(VA)被认为是 Lip降解的最佳底物.在水溶液中,疏水性的木素模型化合物很难被亲水性的 Lip有效降解[4],且酶分子构象容易波动而导致酶活性降低,因此寻求一种新的介质体系来研究 Lip对疏水性化合物的降解具有重要意义.
反胶束是表面活性剂分子在有机溶剂中形成的纳米级分子聚集体.在反胶束体系中,表面活性剂亲水性头基在内形成的极性水核具有增溶生物大分子(如蛋白质、酶等)的作用,外部的有机溶剂有助于疏水性化合物在其中的溶解.因此反胶束成为上述疏水性化合物酶法转化的良好介质.近年来,反胶束体系作为酶催化反应的介质引起了越来越多的关注,但研究的重点却偏向于化学表面活性剂构建的反胶束体系[5-9].同化学表面活性剂相比,生物表面活性剂作为一种环境友好型的天然表面活性剂,具有低毒性、可生物降解性、生态相容性及一定的胶团催化能力等优势[10],而在反胶束酶学中具有较好的应用前景.目前,国内外采用生物表面活性剂构建反胶束体系进行酶催化反应的研究除了本课题组研究报道[11]外,几乎未见其他报道.
本文以 Lip为生物催化剂,对比研究了单鼠李糖脂(RL)反胶束和胶束体系中 Lip催化氧化VA的各主要影响因素,同时探讨了疏水性底物VA在非均相反胶束介质中的分区系数.该研究拓展了胶束酶学中用于构建反胶束体系的表面活性剂的种类,这对于构建适合于胶束酶学研究的新型反胶束体系(生物表面活性剂反胶束酶解体系)而言具有重大意义.
1 材料与方法
1.1 仪器和试剂
岛津 UV-2550型紫外分光光度计(日本Shimadzu公司),TDA-8002电热恒温水浴锅,WHY-2恒温水浴振荡器,HZQ-C空气浴振荡培养箱,TGL-16G离心机,磁力搅拌器.
藜芦醇(VA)购于 Sigma Aldrich公司.异辛烷、正己醇及实验过程中所用的其他试剂均为分析纯,使用过程中无需进一步纯化,水为超纯水.
1.2 鼠李糖脂的生产和纯化
本实验所用的单鼠李糖脂由本课题组制备,其生产和纯化步骤详见文献[12].
1.3 木素过氧化酶(Lip)的生产、分离和纯化
木素过氧化酶(Lip)的生产、分离和纯化步骤详见文献[13].
1.4 反应初速度的测定
1.4.1 含有 Lip酶液的反胶束体系的制备 在小锥形瓶中加入一定体积的异辛烷-正己醇(体积比为1:1)的混合液,按照所需表面活性剂的浓度称取一定量的鼠李糖脂于上述小锥形瓶中,用移液枪加入溶有一定量 Lip的柠檬酸缓冲液(0.1mol/L),pH 值梯度范围为 3.0~4.2,磁力搅拌至形成透明澄清的反胶束体系.相应的胶束体系只需将有机溶剂换成0.1mol/L的柠檬酸缓冲溶液.
1.4.2 反应初速度的测定 VA是黄孢原毛平革菌的一种次生代谢产物,可诱导Lip的合成,是Lip的最适底物.在310nm处无光吸收,而在H2O2的协同作用下,Lip可以将 VA氧化成藜芦醛,产物藜芦醛在 310nm 处有强烈的光吸收[ε=9.3×103L/(mol·cm)][14].测试方法如下:直接称取一定质量的藜芦醇溶于上述反胶束体系中,磁力搅拌均匀后,于30℃下预热2min,取3mL于石英比色皿中,以 10mmol/L的 H2O2溶液引发反应,立即记录310nm处吸光度A随反应时间t的变化曲线.反应初速度ν0[μmol/(L·min)]即为A-t曲线线性部分的斜率.本实验在相同单因子条件下共设置了三组平行试验,试验结果为三次测定的平均值.
2 结果与讨论
2.1 各主要因素对反应初速度的影响
2.1.1 反应体系的 pH值对ν0的影响 反应介质中的pH值是影响酶催化反应的一个重要因素.通过配制一系列不同pH值梯度(3.0~4.2)的柠檬酸缓冲溶液(0.1mol/L),研究pH值对RL构建的反胶束及胶束体系中Lip催化氧化VA反应初速度的影响,其结果如图1所示.一般而言,增溶的缓冲溶液的pH值被认为是反胶束纳米水核的pH值[15].纳米水核的 pH值决定着酶分子的催化构象及其在反胶束体系中的增溶能力,同时 pH值的变化还可能改变酶分子与反胶束液膜之间的相互作用[16].
如图 1所示,反胶束体系中催化反应的最适pH值为3.8,这比在胶束水溶液(3.4)中的略高,这可能和Lip的结合和氧化位点有关.Lip的结合和氧化位点暴露于酶蛋白的表面,当 Lip增溶到反胶束中后,其表面暴露的位点很容易受到 RL内表面极性头基的影响[17].由于反胶束反应体系中的 pH 值低于 Lip的 pI:4.2~4.9[18],从而使得 Lip带正电荷,这样 RL液膜内表面带负电荷的阴离子头基和带正电的 Lip之间产生有利的静电相互作用使得 Lip暴露的位点可以耐受相对较大的pH值.

图1 pH值对鼠李糖脂反胶束和胶束体系中Lip氧化VA反应初速度的影响Fig.1 Effect of pH on the initial reaction rate of VA oxidation by Lip hosted in the RL reverse micellar and micellar systems
2.1.2 表面活性剂溶度[RL]对ν0的影响 只有当表面活性剂浓度达到临界胶束浓度(cmc)时才能形成反胶束[19],单鼠李糖脂在水相及有机相中的临界胶束浓度分别为0.012, 0.055mmol/L[20].依据上述cmc值,对比了两种体系中[RL]对Lip催化VA反应初速度的影响.由图2可以看出,固定w0(c[水]/c[表面活性剂],无量纲)为 15.0 时,在 RL构建的反胶束体系中Lip催化氧化VA的反应初速度随着RL浓度的增加出现类似钟型的变化趋势.[RL]在1~10cmc之间Lip的活力几乎为零.在100~180cmc(约 5.5~10mmol/L)的范围内,随着[RL]的增大,催化活力逐渐增大.最大酶活出现在[RL]=10mmol/L处,超过 10mmol/L时随着[RL]的增加酶活迅速降低.
在w0值恒定时,表面活性剂浓度的增加只会引起反胶团个数的增加而不会影响反胶团体积的大小,开始阶段由于反胶团个数的增加增大了酶分子与底物的接触和碰撞机率,因此反胶束酶系统中催化反应速率随着表面活性剂浓度的增加出现增大的趋势;超过一定值后,随着RL浓度的增加,酶分子在反胶团中的浓度降低,单位时间内酶和底物的接触机率变小从而又引起酶活的下降[21].而在鼠李糖脂构建的胶束体系中 Lip催化VA反应除了在低浓度30μmol/L下对Lip有微弱的激活作用外,从图形的整体趋势来看,胶束体系中Lip催化活力随着RL浓度的增加而降低.对比可知,在胶束体系中较低的表面活性剂浓度即表现出对 Lip催化活性的强烈抑制作用,这可能和水溶液中表面活性剂对酶蛋白的变性作用有关.在胶束体系中,Lip分散于水溶液中,酶分子容易受到表面活性剂单体的变性作用,从而改变其催化构象,导致催化活力的降低[22].

图2 RL浓度对反胶束和胶束体系中Lip催化氧化VA反应初速度的影响Fig.2 Effect of the concentration of RL on the initial reaction rate of Lip-catalyzed oxidation VA in the reverse micellar and micellar systems
2.1.3 含水率w0对ν0的影响 含水率w0直接影响反胶束纳米水核大小,进而影响反胶束增溶能力和酶在其中的催化效率[23].如图 3所示,Lip催化氧化VA的反应初速度随着w0的变化呈现近似钟型的变化趋势,w0在6.0以下Lip基本无催化活性(图中略去),在w0为 6.0~15.0时,随着w0的增加催化反应初速度逐渐增大,w0=15.0时催化速率达到最大值,之后随着w0的增加催化速率几乎成线性减小.
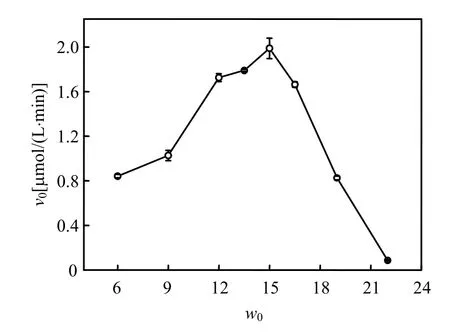
图3 w0对RL反胶束体系中Lip氧化VA反应初速度的影响Fig.3 Effect of w0 on the initial reaction rate of VA oxidation by Lip entrapped in the RL reverse micellar system
处于最佳含水率时,反胶束纳米水核空间的大小与酶分子大小相当,酶分子在反胶束中可以保持最佳的构象,从而表现出较高酶活;当w0小于此值时,反胶束水核体积较小,不能很好地增溶酶分子,致使酶分子过多地暴露于有机溶剂中,从而使酶的催化活性降低;而当w0大于此值时,反胶束水核尺寸大于酶分子大小,酶分子在纳米水核中的自由度增加,其构象受到扰动而变得松散.只有在最佳含水量时酶蛋白结构的动力学刚性和热力学稳定性之间达到最佳平衡点,酶才表现出最大活力[24].
2.2 Lip在两种介质中的活性和稳定性
由于酶在增溶进反胶束的过程中其活性和稳定性会随之改变[25],为此,实验测定并讨论了Lip酶在RL胶束和反胶束体系中的催化活性和酶活稳定性.胶束体系的实验条件:pH3.4,[RL]=0.012mmol/L,[H2O2]=2.45mmol/L;反胶束体系的实验条件:pH3.8,[RL]=10mmol/L,w0=15.0,[H2O2]=74μmol/L.由图4可知,在RL胶束介质中Lip酶活在8h内损失超过50%,10h后Lip基本完全失活,而在RL反胶束体系中Lip的半衰期达到40h左右,在62h时仍然保持原有酶活的32%;此外,RL构建的反胶束体系中Lip的最大催化活力比其在胶束水溶液中高了近2.86倍,表现出超活性.

图4 Lip在两种介质中的活性和稳定性Fig.4 Activity and stability of Lip in the two medium

图5 H2O2浓度对RL反胶束及胶束体系中Lip催化氧化VA反应初速度的影响Fig.5 Effect of the H2O2 concentration on the initial reaction rate of Lip-catalyzed oxidation VA in the RL reverse micellar and micellar system
在最优催化条件下,酶在两种体系中的活性和稳定性产生较大差别的原因较为复杂,可能是因为RL反胶束中的纳米水核模拟了生物细胞内的微环境,反胶束的存在使得增溶于纳米水核中的 Lip与有机相彼此分离,避免了酶与有机溶剂的直接接触,有效地保护了酶的活性部位,对其催化构象的变化有一定程度的束缚作用,从而提高了酶的催化性能和稳定性;而在胶束体系中,Lip分散于水溶液中,酶分子刚性减弱、波动性变大,从而导致了活性降低[26-27].Kimura等[28]证实在AOT反胶束体系中, Lip酶由于受到反胶束纳米水核的保护而导致 Lip的活性明显增强,类似现象出现在反胶束体系中锰过氧化物酶(Mnp)[29]、漆酶(Lac)[30]、纤维素酶(Cellulose)[31]、碱性磷酸酶(pNPPsase)[32]等的催化特性研究中.
2.3 H2O2抑制浓度和底物分区系数P的确定
2.3.1 H2O2抑制浓度的确定 对于Lip催化氧化VA的反应而言,H2O2既是反应的启动剂又是反应的抑制剂[33],因此在讨论胶束和反胶束中的反应动力学机制时确定H2O2的抑制浓度十分必要.图5表示在两种介质中反应初速度随着H2O2浓度的变化曲线.可以看出,反胶束体系中Lip的催化活性在很大程度上取决于 H2O2的浓度.当H2O2浓度为74μmol/L时反应初速度达到最大值,当H2O2浓度达99μmol/L时即表现对酶活的强烈抑制作用,因此动力学实验中 H2O2浓度范围应选择在34~74μmol/L之间.然而在RL胶束水溶液中当H2O2的浓度达到3.25mmol/L时Lip仍具有催化活性.在反胶束介质中 H2O2的抑制浓度远低于水溶液中,主要是由反胶束介质的微观不均一性引起的.也就是说,在反胶束体系中,H2O2主要增溶于纳米水核中,这高度浓缩了反应过程中H2O2的浓度,从而使 H2O2在较低的浓度表现出对酶活的强烈抑制作用[34].

图6 在一组不同θ下,在RL反胶束体系中Lip催化氧化VA反应的初速度对VA浓度的双倒数图Fig 6 Double reciprocal plot of the initial rate of Lipcatalyzed oxidation VA versus the concentration of VA at several different values of θ in the RL reverse micellar system
2.3.2 VA在反胶束体系中分区系数P的确定 根据两相模型(biphasic model)[35],将鼠李糖脂/异辛烷-正己醇/水反胶束体系看成由有机相和反胶束拟相两部分组成.亲水性的Lip酶增溶于反胶束拟相中,疏水性的底物VA在有机相和反胶束拟相中均有较大的溶解度[34].因此在讨论 RL反胶束体系中Lip酶的催化动力学时应首先考虑VA在两相中的分配系数P.由两相模型(底物分区考虑在内):

其中:kcat,app=kcat,mic

分配系数P定义如下:
式中:[E]0,app,[S]0,app为酶和底物的表观初始浓度;kcat,app,kcat,mic为表观催化常数和反胶束拟相催化常数;km,app,km,mic为表观米氏常数和反胶束拟相米氏常数;θ为反胶束体系中水的体积分数;P为底物在反胶束拟相和有机相中的浓度比; [S]mic,[S]os为底物在反胶束拟相和有机相中的浓度.
由式(1)得:
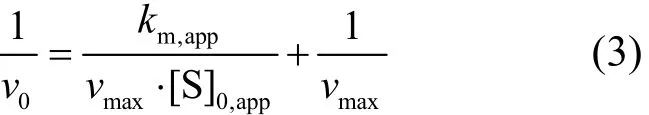
由式(2)得:
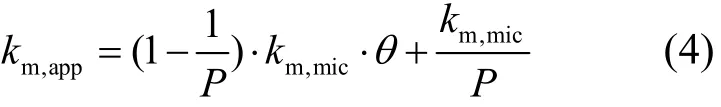
由式(4)可知,在一组固定的θ下,产生几个固定的km,app,此时式(3)中v0-1~[S]0,app-1双倒数图为一系列有着相同截距vmax-1,不同斜率km,app/vmax的直线.二级图km,app~θ仍为一直线,其斜率与截距之比为P-1,据此可以求出分配系数P.
图6表示在几个固定的θ下,反应初速度ν0对不同浓度的 VA的双倒数曲线.由图 6可知ν0-1~[VA]0-1双倒数曲线皆可拟合成较好的直线,且在不同θ下,直线相交于y轴上同一点.图6中斜率km,app/vmax与截距vmax-1之比km,app对θ二次作图,得一直线(图7).直线斜率为539,截距为7.77,则有斜率/截距=69.4,即P-1=69.4,据此可算出P=70.4.

图7 km,app对θ二次作图Fig.7 Replot of the values of km,app versus θ
3 结论
3.1 在RL构建的反胶束体系中 Lip催化氧化VA受反胶束纳米水核pH值、含水率w0和鼠李糖脂浓度等主要因素的影响,最佳催化活力出现在:pH=3.8,w0=15.0,[RL]=10mmol/L处,处于最适条件下,Lip的半衰期长达40h,对比Lip在反胶束介质和胶束介质中的催化活性可知,Lip在RL反胶束体系中表现出了超活性.
3.2 反胶束的纳米水核高度浓缩了反应过程中H2O2的浓度,从而使 H2O2在较低的浓度(99μmol/L)时即表现出对酶活的强烈抑制作用,因此在后续研究RL反胶束体系中Lip催化氧化VA的动力学机制时应该选取 H2O2的浓度范围为 34~74μmol/L 之间.
3.3 在鼠李糖脂构建的反胶束体系中,疏水性底物 VA主要增溶于反胶束拟相中,根据两相模型可计算其在反胶束拟相和有机溶剂相间的分区系数为 70.4.分区系数的确定对于进一步讨论RL反胶束体系中Lip催化氧化VA的动力学机制而言具有重要意义.
[1]田朝光,马延和.真菌降解木质纤维素的功能基因组学研究进展[J]. 生物工程学报, 2010,26(10):1333-1339.
[2]宋安东,薛仁军,谢 慧,等.不同微生物降解木质纤维素效率和过程的对比研究 [J]. 安全与环境学报, 2009,9(2):1-6.
[3]Tien M, Kirk T K.Lignin-degarding enzyme from the hymenomycetePhanerochaete chrysosporiumburds [J]. Science, 1983,221:661-663.
[4]Okazaki S,Goto M,Furusaki S,et al.Preparation and catalytic per-formance of surfactant-manganese peroxidase-MnⅡternary complex in organic media [J]. Enzym. Microb. Technol., 2001,28:329-332.
[5]姬广磊.Irpex lacteusdft-1漆酶在反胶束中的催化性能研究 [J].环境科学与技术, 2009,32(12):79-84.
[6]Andrade S M, Carvalho T I, Viseu M I, et al. Conformational changes ofβ-lactoglobulin in sodium bis(2-ethylhexyl) sulfosuccinate reverse micelles [J]. Eur. J. Biochem., 2004,271: 734-744.
[7]张文娟,王 丹,黄锡荣,等.非离子反胶束中木素过氧化物酶催化性能研究 [J]. 生物工程学报, 2005,21(4):654-657.
[8]Mitra R N, Dasgupta A, Das D, et al. Geometric constraints at the surfactant headgroup: effect on lipase activity in cationic reverse mi –celles [J]. Langmuir, 2005,21:12115-12123.
[9]伏振宇,李志光,何纯莲.反胶束体系中固定化胃蛋白酶的催化性质研究 [J]. 化学与生物工程, 2011,28(10):75-78.
[10]Xie Y W, Ye R Q, Liu H L. Synthesis of silver nanoparticles in reverse micelles stabilized by natural biosurfactant [J]. Colloids and Surfaces A: Physicochem. Eng. Aspects, 2006,279:175-178.
[11]王伟伟,袁兴中,曾光明,等.逆胶束体系中纤维素酶解特性研究[J]. 环境科学, 2010,31(9):2202-2207.
[12]Liang Y S, Yuan X Z, Zeng G M, et al. Biodelignification of rice straw byPhanerochaete chrysosporiumin the presence of dirhamn –lipid [J]. Biodegradation, 2010,21(4):615-624.
[13]李越中,高培基,王祖农.黄孢原毛平革菌合成木素过氧化物酶的营养调控 [J]. 微生物学报, 1994,34(1):29-36.
[14]王 丹,黄锡荣,宋少芳,等.非离子表面活性剂存在下过氧化氢对木素过氧化物酶的抑制动力学研究 [J]. 山东大学学报(理学版), 2003,38(2):108-111.
[15]陈建波,夏春谷,李树本,等.反胶束对辣根过氧化物酶催化反应的影响 [J]. 分子催化, 1999,13(6):453-456.
[16]刘伟东,聂开立,鲁吉珂,等.反胶束体系中脂肪酶催化合成生物柴油 [J]. 生物工程学报, 2008,24(1):142-146.
[17]Johjima T, Wariishi H, Tanaka H. Veratryl alcohol binding sites of lignin peroxidase fromPhanerochaete Chrysosporium[J]. J. Mol.Catal. B-Enzym., 2002,17:49-57.
[18]杨金水,袁红莉,刘庆洪,等.斜卧青霉菌P6木素过氧化物酶的纯化与特性 [J]. 中国农业大学学报, 2004,9(2):1-5.
[19]Tonova K, Lazarova Z. Reversed micelle solvents as tools of enzyme purification and enzyme-catalyed conversion [J]. Biotechnol.Adv., 2008,26:516-532.
[20]崔凯龙,袁兴中,曾光明,等.生物表面活性剂用于逆胶束体系的构建及微水相条件的优化 [J]. 中国环境科学, 2011,31(9):1444-1450.
[21]Janina R N, Masaaki I. Effect of AOT on enzymatic activity of the organic solvent resistant tyrosinase fromStreptomycessp.REN-21 in aqueous solutions and water-in-oil microemulsions [J].J. Colloid Interf. Sci., 2005,284:674-679.
[22]李学刚.表面活性剂的疏水性及电性对肌酸激酶的活力及复性能力的影响 [J]. 生物物理学报, 1992,8(4):596-600.
[23]Michizoe J, Goto M, Furusaki S. Catalytic activity of lactase hosted in reversed Micelles [J]. J. Biosci. Bioeng., 2001,92(1):67-71.
[24]王元鸿,褚 莹,刘景林,等.反胶束体系中脂肪酶催化合成异丁酸异戊酯 [J]. 高等学校化学学报, 2004,25(9):1684-1688.
[25]Marhuenda-Egea F C, Piera-Velázquez S, Cadenas C, et al.Kinetic studies of an extremely halophilic enzyme entrapped in reversed micelles [J]. Biocatal. Biotransform., 2000,18(3):201-222.
[26]María A B, Elsa B A, Junan J S, et al. Kinetics of reactions catalyzed by enzymes in solutions of surfactants [J]. Adv. Colloid Interf. Sci., 2008,136:1-24.
[27]Martinek K, Klyachko N L, Kabanov A V, et al. Micellar enzymelogy: Its relation to membranology [J]. Biochim. Biophys. Acta,1989, 981(2):161-172.
[28]Kimura M, Michizoe J, Oakazaki S, et al.Activation of lignin peroxidase in organic media by reversed micelles [J]. Biotechnol.Bioeng., 2004,88(4):495-501.
[29]Michizoe J, Uchimura Y, Ichinose H, et al. Activation of manganese peroxidase in an organic medium using a mediator [J].Biochem. Eng.J., 2004,19:43-46.
[30]Michizoe J, Ichinose H, Kamiya N, et al. Biodegradation of phenolic environmental pollutants by a surfactant-laccase complex in organic media [J]. J. Biosci. Bioeng., 2005,99(6):642-647.
[31]Chen N, Fan J B, Xiang J,et al.Enzymatic hydrolysis of microcrystalline cellulose in reverse micelles [J]. Biochim. Biophys. Acta,2006,1764:1029-1035.
[32]Marhuenda-Egea F C, Piera-Velázquez S, Cadenas C,et al. Stability of an extreme halophilic alkaline phosphatase from Halobacterium salinarium in non-conventional medium [J]. J. Biotechnol.,2001,87:255-261.
[33]Liu J T, Huang X R, Gao P J.Kinetic studies on the lignin peroxidase catalyzed oxidation of veratryl alcohol by H2O2in AOT/iso-octane/toluene/water reverse micelles [J]. Chinese J.Chem., 2007,25:1627-1631.
[34]Zhang W J, Huang X R, Li Y Z, et al. Catalytic activity of lignin peroxidase and partition of veratryl alcohol in AOT/isooctane/tolu-ene/water reverse micelles [J]. Appl. Microbiol. Biotechnol.,2006,70:315-320.
[35]Lissi E A,Abuin E B.A general treatment for meaningful comparison of rate parameters of enzyme-catalyzed reactions in aqueous and reverse micellar solutions [J]. Langmuir, 2000,16,10084-10086.
