葡萄糖“糖酵解-有氧氧化脱耦联”在慢性心肌肥厚进展中的作用
2013-08-28周和平朱海龙顾春虎陈涛熊红燕孙国成
周和平,朱海龙,顾春虎,陈涛,熊红燕,孙国成
目前慢性心力衰竭的发病率及病死率均较高[1-2],且机制仍不明确。能量代谢异常与心力衰竭的相关性以及两者之间可能存在的因果关系近年来受到广泛关注,但目前尚无资料表明哪种纠正代谢异常的治疗措施具有确切的疗效。一般认为,在中晚期或终末期心力衰竭时,脂肪酸的氧化代谢显著下调,心肌更多地利用葡萄糖作为代谢底物[2];在肥厚的心脏中存在葡萄糖利用及糖酵解过程增强﹑有氧氧化过程相对减弱,即“糖酵解-有氧氧化脱耦联”现象[3],但其具体的病理生理意义仍不清楚。二氯乙酸(dichloroacetate,DCA)是一种特异作用于线粒体的小分子化合物,可通过抑制丙酮酸脱氢酶激酶活性激活丙酮酸脱氢酶(PDH),增强线粒体的氧化磷酸化,从而改善胞质内糖酵解和线粒体内有氧氧化之间的脱耦联[4],但有关其改变葡萄糖代谢途径后对心脏的结构和功能有何影响仍不明确。本研究旨在明确葡萄糖“糖酵解-有氧氧化脱耦联”在慢性心力衰竭进展过程中的作用,进一步深入认识葡萄糖代谢在心力衰竭病程中的作用及机制,以期从改善代谢的角度为防治心力衰竭提供新策略和新靶点。
1 材料与方法
1.1 实验动物﹑试剂及仪器 鼠龄10周左右的雄性近交系C57BL/6J小鼠62只,新生Wistar大鼠(1~3d)15只。所有动物实验均经第四军医大学动物伦理委员会批准。低糖DMEM培养基﹑Percoll分离液﹑胎牛血清(Gibco BRL公司),抗磷酸化丙酮酸脱氢酶(p-PDH)抗体(Calibio公司),抗PDH抗体(MitoSciences公司),抗GAPDH抗体(Cell signaling公司),二氯乙酸(DCA,Sigma公司)。体视显微镜(Leica公司),vevo770超声分析系统(Visual Sonics公司),LAS-3000化学发光成像系统(富士公司)。
1.2 慢性心力衰竭模型的建立及实验分组 本研究采用国际上通用的主动脉缩窄(transaortic constriction model,TAC)模型[5]作为左室压力超负荷模型。小鼠麻醉后行气管插管﹑接小型动物呼吸机,呼吸支持频率40次/min,用1%~2%的异氟烷维持麻醉。从小鼠左侧第2肋间进胸,在体视显微镜下分离主动脉弓,在左右颈动脉之间分离横主动脉,放置27号针头,用10-0尼龙线打结后迅速抽出针头。检查无出血后,分别缝合肌层与皮肤。假手术组处理同上,但不进行结扎。动物分组:①将10只正常小鼠分为DCA处理组(n=5)及对照组(n=5),以观察DCA处理对正常小鼠心肌组织p-PDH表达的影响。②将假手术组及TAC术后3d﹑2周﹑20周不同时间点各4只(共16只)小鼠用于分析TAC术后p-PDH的动态表达变化;将12只小鼠分为假手术组﹑TAC组及TAC+DCA处理组(160mg/kg[4]加入饮水中),每组4只,用于分析DCA处理对TAC术后第3天p-PDH表达的影响。③另将24只TAC小鼠分为DCA处理组(160mg/kg[4]加入饮水中,n=12)及对照组(正常饮水,n=12),以观察DCA处理对TAC模型小鼠心功能和生存率的影响。
1.3 超声心动图检测 小鼠麻醉后仰卧位固定于vevo770超声分析系统操作台上,用1%~2%的异氟烷维持麻醉。先从主动脉弓切面证实TAC模型,彩色多普勒分析缩窄处最大血流速度和压差,将缩窄率为<60%或>80%的动物筛除(与国际报道的70%左右的狭窄率保持一致)。维持小鼠心率在450次/min左右,采用美国超声协会标准的前缘标准测量左室收缩末期和舒张末期内径(LVESD和LVEDD),左室收缩功能射血分数(EF)按下列公式计算:EF(%)=[(LVEDV–LVESV)/LVEDV]×100。
1.4 Western blotting检测p-PDH及PDH的表达 分别于TAC术后3d﹑2周﹑20周将小鼠脱颈处死,即刻取出心脏,将心肌组织用预冷至0℃的PBS(pH 7.4)清洗后移入预冷的裂解液(Tris·HCl 20mmol/L﹑NaCl 50mmol/L﹑NaF 50mmol/L﹑Na4P2O750mmol/L﹑Sucrose 250mmol/L﹑Na3VO42mmol/L﹑DTT 1mmol/L和1%复合型蛋白酶抑制剂)中并剪碎。组织匀浆后于4℃ 12 000×g离心10min,取上清;采用BCA蛋白定量试剂盒进行蛋白定量,样品行SDSPGEA电泳,采用半干电转法转移到PVDF膜上,5%脱脂奶粉室温封闭1h;将PVDF膜和一抗(PDH和p-PDH单抗)一起预处理,4℃过夜;用PBST缓冲液漂洗,将膜与辣根过氧化物酶标记的二抗在37℃预处理1h;洗去所有未结合的二抗,使用ECL-plus免疫检测试剂盒进行化学发光检测。
1.5 新生大鼠心肌细胞的分离培养 采用新生Wistar大鼠(1~3d),开胸摘取心脏后放入盛有预冷(4℃)的无钙镁PBS平皿中洗去血凝块和红细胞,并剪去心房,将心肌剪成约1mm3大小的碎块,放入三角烧瓶中;加入10倍体积0.05%的胰酶,在磁力搅拌器上磁力消化(100r/min×10min,35~37℃);消化完后用吸管轻轻吹打,静置2min,自然沉淀,弃上清(上清液内主要含红细胞﹑细胞碎屑及死亡细胞);再次消化,反复2次后,取上清。将上清移入无菌离心管中,加入4℃预冷的含小牛血清的DMEM终止胰酶作用,轻吹﹑混匀后,1000r/min×10min﹑4℃离心,弃上清,反复3次,以洗去残存胰酶。在各次消化所得的细胞沉淀中加入培养液,集中混匀制成悬液,经细胞计数后,用含10%小牛血清的DMEM液接种培养,24h换液1次,48h后改用无血清的DMEM液继续培养24h。
1.6 DCA处理对心肌细胞p-PDH表达的影响 分别用5﹑10﹑30mmol/L的DCA与心肌细胞共同孵育,3h后提取细胞总蛋白行Western blotting检测分析p-PDH表达的变化,同时按照试剂盒说明书行免疫细胞荧光染色。
1.7 TUNEL法原位检测细胞凋亡 操作按试剂盒说明书进行。心肌组织常规石蜡包埋,每一个标本取3个不同部位切片,厚4μm。心肌细胞核用DAPI衬染呈蓝色,凋亡细胞呈深浅不一的绿色颗粒状。在计算机图像分析仪上,在400倍视野下,每张切片拍摄5个阳性视野,每个视野计数200个细胞核,阳性细胞所占的百分比作为凋亡细胞阳性指数(AI)。AI=凋亡阳性细胞数/总细胞数×100。AI作为心肌细胞凋亡数量的半定量参数。
1.8 统计学处理 采用SPSS 17.0进行统计学分析,数据以±s表示,两组间的比较采用非配对t检验(unpaired Student's t-test),多组间比较采用单因素方差分析(ANOVA)分析,P<0.05为差异有统计学意义。
2 结 果
2.1 小鼠TAC模型的超声分析结果 超声心动图分析进一步证实,手术狭窄能稳定地控制在70%左右,图1左侧血流频谱显示最大血流速度在3.5~4.0m/s,右侧彩色多普勒测得跨狭窄处压力差50~60mmHg,且在狭窄处可见明显的五彩血流信号(图1)。
2.2 慢性心力衰竭进展过程中p-PDH的动态表达采用TAC模型小鼠分析慢性心力衰竭进展过程中
p-PDH表达的动态变化,并以此间接反映糖酵解和氧化磷酸化的相对水平(p-PDH的表达与糖酵解水平呈正相关,与氧化磷酸化水平呈负相关)。Western blotting结果显示,小鼠TAC模型中p-PDH的表达在术后第1天即开始升高,在术后第3天达到峰值,术后2周仍升高,而在术后20周时表达很低(图2)。
2.3 DCA对p-PDH表达的影响 体外实验证明药物DCA可以使培养的心肌细胞p-PDH表达降低且呈剂量依赖性,并在10.0mmol/L时达到最大效应,免疫细胞荧光染色进一步证明了DCA的作用(图3)。体内实验进一步证明,并在正常小鼠中,DCA处理可增强有氧氧化即p-PDH表达降低;在TAC模型小鼠中,DCA可改善慢性心肌肥厚过程中的糖酵解增强﹑有氧氧化降低(图4),从动物水平证明了DCA(160mg/kg)的有效性。
2.4 DCA处理对TAC小鼠心功能的影响 用DCA药物被动地改善“糖酵解-氧化磷酸化脱耦联”﹑加强线粒体氧化磷酸化并不能改善肥厚心脏的心功能,相反却加速了心功能的恶化,TAC术后4﹑10周,DCA处理组心功能明显差于TAC模型组(图5﹑6),进而小鼠20周生存率降低(图7)。
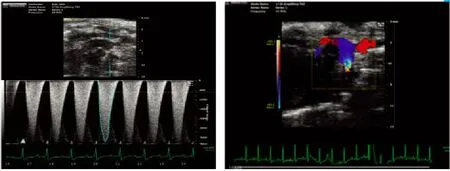
图1 小鼠TAC模型的超声分析Fig.1 Echocardiographic analysis of TAC model of mice

图2 TAC小鼠p-PDH水平的动态变化Fig.2 Dynamic expression of p-PDH in TAC mice
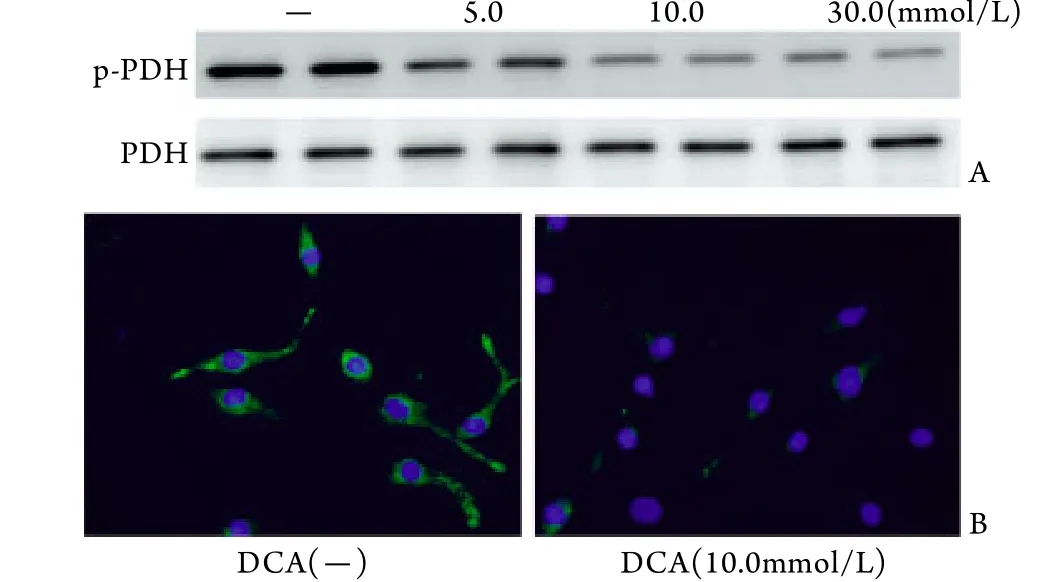
图3 DCA对心肌细胞p-PDH表达水平的影响Fig.3 Effect of DCA on cardiomyocyte p-PDH expression A. Western blotting result; B. Immunofluorescent staining
2.5 DCA处理对心肌细胞凋亡的影响 TUNEL法半定量分析结果显示,术后DCA处理组的凋亡指数(29.3±2.5)明显高于单纯TAC组(18.8±1.9,P<0.05),且两组均明显高于假手术组(9.1±1.5,P<0.05,图8)。

图4 DCA对小鼠PDH及p-PDH蛋白表达的影响Fig.4 Effect of DCA on mice p-PDH and PDH expression A. Normal mice; B. TAC mice
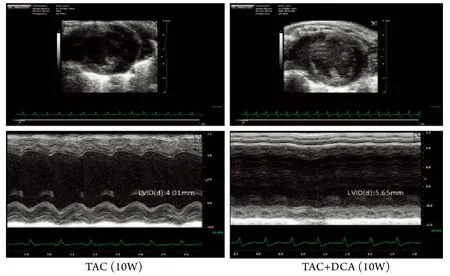
图5 TAC小鼠超声心功能分析Fig.5 Cardiac function evaluation by echo in TAC mice

图6 TAC小鼠EF的动态变化Fig.6 Dynamic analysis of EF in TAC mice
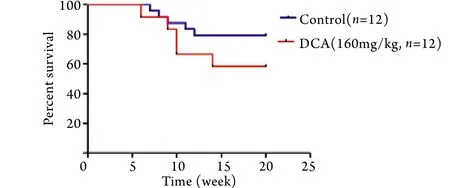
图7 TAC小鼠生存曲线Fig.7 Survival curve of TAC mice
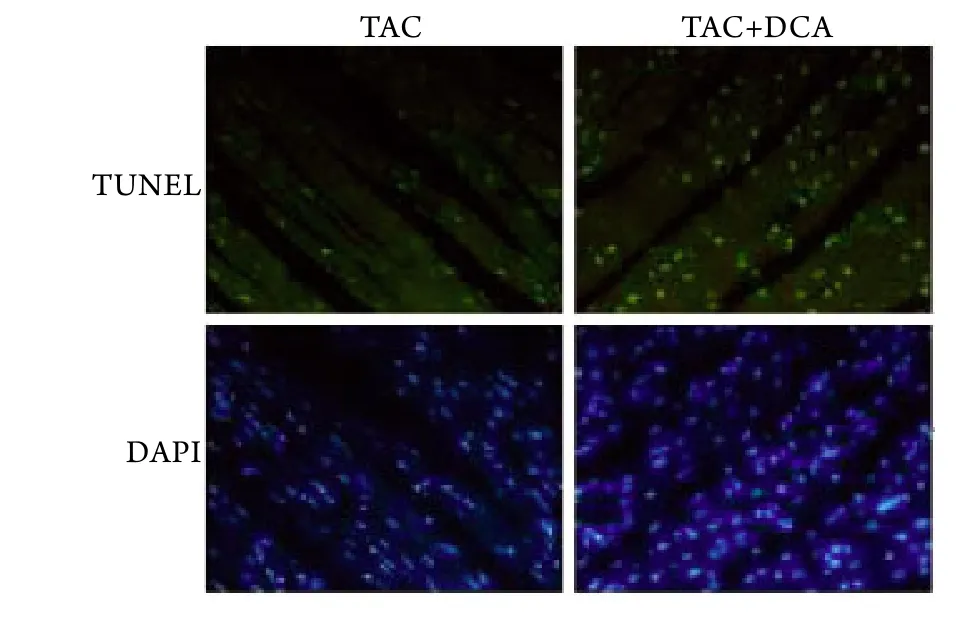
图8 心肌细胞凋亡(DAPI ×40)Fig.8 Apoptosis of cardiomyocyte (DAPI ×40)
3 讨 论
已有文献表明,在肥厚的心脏中存在葡萄糖利用及糖酵解过程增强﹑有氧氧化过程相对减弱的现象,这就是所谓的“糖酵解-有氧氧化脱耦联”[3],但其具体的病理生理意义仍不明确。因糖酵解ATP生成效率远较有氧氧化为低,因此通常认为“糖酵解-有氧氧化脱耦联”因能量供应少而对心功能不利。然而,我们通过压力超负荷模型和高血压大鼠模型首次发现,“糖酵解-有氧氧化脱耦联”在慢性心力衰竭的进展中可发挥代偿性保护作用,而促进糖酵解向有氧氧化转化则加重心功能障碍,提示糖代谢异常对心力衰竭发展影响的新机制,与先前报道的文献中右室肥厚中的变化规律一致[6]。
针对上述问题,我们想到在肿瘤细胞中存在着一个有趣的现象“Warburg效应”。根据此效应,在肿瘤细胞和增殖细胞中,无论氧供充足与否,大部分(85%左右)的葡萄糖都经由丙酮酸代谢为乳酸,即“有氧糖酵解”;而在正常已分化组织中,在氧供充足时主要在线粒体内进行氧化磷酸化,只有在缺氧应激时葡萄糖才被代谢为乳酸,称为“无氧糖酵解”[7]。肿瘤细胞采用这种相对低效率的ATP生成方式来提供自身代谢所需能量的可能原因有两个:第一,补充用于细胞增殖所必需的生物大分子如核苷酸﹑氨基酸等;第二,减少线粒体氧化磷酸化过程活性氧自由基(ROS)的生成,线粒体氧化磷酸化是ROS生成的主要来源。另外,过多的ATP和柠檬酸会抑制糖酵解﹑减少磷酸戊糖代谢途径的流量,从而使NADPH的生物合成减少,而NADPH是谷胱甘肽氧化还原循环的重要辅助因子,从而加重细胞的氧化损伤。因此可以推断,肿瘤细胞采用有氧糖酵解作为能量代谢的方式主要是满足增殖时对原材料的需要以及增强自身对外界各种不良应激的抵抗性。
与肿瘤细胞的糖代谢状况类似,在心力衰竭进展的各个时期,均有心肌能量代谢的变化。因为心室重构使单位重量的心肌毛细血管数目减少,氧的弥散间距增大,故心肌细胞缺氧。同时在肥厚心肌存在“糖酵解-有氧氧化脱耦联”,葡萄糖供能效率降低,这就加重了心肌的缺氧。慢性心力衰竭时多种因素导致氧化代谢受损,氧自由基生成增多,体内活性氧堆积,形成氧化应激状态。本研究证实,增强葡萄糖的有氧氧化,可能通过诱导氧化应激导致心肌细胞凋亡,而细胞凋亡与心脏重塑密切相关,心肌细胞凋亡很可能是有氧氧化增强使慢性心肌肥厚从“代偿”向“失代偿”转折的关键因素[8]。
综上所述,本研究结果发现,“糖酵解-有氧氧化脱耦联”在慢性心力衰竭的进展中可发挥代偿性的保护作用;心肌的葡萄糖代谢从糖酵解向氧化磷酸化转变促进了代偿性心肌肥厚向失代偿性心力衰竭的转变;葡萄糖氧化磷酸化促进心力衰竭的机制与ROS生成增多从而导致心肌细胞凋亡增多有关。本研究结果揭示了心力衰竭失代偿的一个新的调节机制,可能为临床从改善代谢的角度防治和延缓心力衰竭提供新策略和新靶点。
[1] McMurray JJ, Pfeffer MA. Heart failure[J]. Lancet, 2005, 365:1877-1889.
[2] Shen M, Liu B, Wang HC, et al. Observations on short-term curative effect of resynchronization therapy for patients with chronic heart failure[J]. Med J Chin PLA, 2008, 33(8): 1016-1018. [沈敏, 刘兵, 王海昌, 等. 心脏再同步化治疗慢性心衰的短期疗效观察[J]. 解放军医学杂志, 2008, 33(8): 1016-1018.]
[3] Neubauer S. The failing heart-an engine out of fuel[J]. N Engl J Med, 2007, 356(11): 1140-1151.
[4] Lydell CP, Chan A, Wambolt RB, et al. Pyruvate dehydrogenase and the regulation of glucose oxidation in hypertrophied rat hearts[J]. Cardiovasc Res, 2002, 53(4): 841-851.
[5] Stacpoole PW, Kerr DS, Barnes C. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children[J]. Pediatrics, 2006, 117(5): 1519-1531.
[6] Rockman HA, Ross RS, Harris AN, et al. Segregation of atrialspecificand inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy[J].Proc Natl Acad Sci USA, 1991, 88: 8277-8281.
[7] Piao L, Fang YH, Cadete VJ, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle[J]. J Mol Med, 2010, 88(1): 47-60.
[8] Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation[J]. Science, 2009, 324(5930): 1029-1033.
[9] Wu WK, Yang H, Zhao MQ. Mechanisms of heart failure induced by adriamycin[J]. Chin J Pathophysiol, 2004, 20(8): 1437-1439.[吴伟康, 杨辉, 赵明奇. 阿霉素性心力衰竭模型的氧化应激和凋亡机制[J]. 中国病理生理杂志, 2004, 20(8): 1437-1439.]
