云南松松塔中planchol E的手性结构与光谱的密度泛函研究
2013-08-16刘光明莽朝永
杨 颖,刘光明,雷 婷,莽朝永,2*
(1.大理学院药学与化学学院,云南大理 671000;2.大理学院药物研究所,云南大理 671000)
云南松松塔中planchol E的手性结构与光谱的密度泛函研究
杨 颖1,刘光明1,雷 婷1,莽朝永1,2*
(1.大理学院药学与化学学院,云南大理 671000;2.大理学院药物研究所,云南大理 671000)
立体化学结构与手性光谱关系研究,对手性化合物在手性材料和手性药物等方面的应用,是前期性的基础工作。对一种分离自云南松松塔的手性苯酚类化合物(planchol E),进行了密度泛函理论计算研究。计算结果表明:测定的NRM谱主要体现了异构体1的特征,而测定IR谱主要是异构体2的。由于异构体2有分子内氢键,导致两种异构体的光谱差异很大。
planchol E;密度泛函方法;电子圆二色谱;振动圆二色谱;氢键
在云南省西北部,云南松(Pinus yunnanensis)是一种重要的资源植物,其化学成分具有抗癌和抗HIV-1活性。近年来,从云南松中已分离出了多种化合物,包括萜类、黄酮类、木质素类和生物碱类物质〔1-2〕。我们从云南松松塔中分离到一种罕见的具有手性结构的苯酚化合物〔3〕,命名为planchol E,分子骨架是刚性的,由一个苯酚环、一个含氧六元环、两个含氧五元环融合而成。见图1。实验上测定了该化合物的旋光度、核磁共振谱、红外振动光谱和紫外电子吸收光谱。
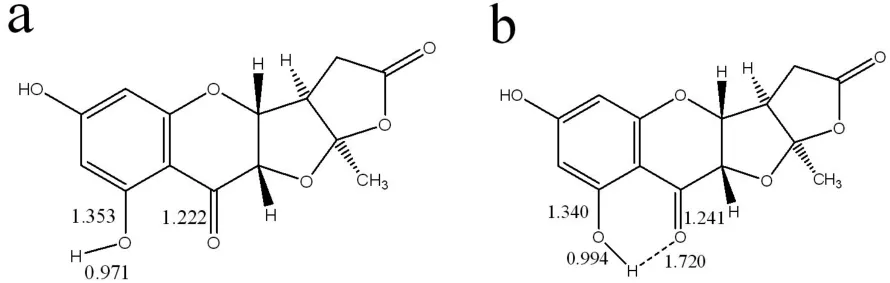
图1 化合物1分子结构:异构体1(a)和异构体2(b)
手性化合物的不同对映异构体,不但具有不同的手性光谱,而且常会表现出不同的生物活性。手性分子的立体结构与手性光谱的关系研究,对研究手性材料和开发手性药物,是一项前期性的基础工作。在目前的工作中,我们采用密度泛函(DFT)方法,优化了planchol E的分子结构,并计算了核磁共振谱(NRM)、紫外电子吸收光谱(UV)和电子圆二色谱(ECD)、红外振动吸收光谱(IR)和振动圆二色谱(VCD)。由于酚类化合物含有活泼的羟基而具有抗氧化活性,对人体健康起到有益作用,是许多饮料如茶、葡萄酒和咖啡的主要成分,具有抗炎症、抗病毒、抗变态、抗肿瘤等作用,对其分子结构和光谱特征的深入研究,对这类化合物在手性药物方面的应用具有重要意义。
1 计算方法
根据图1,planchol E有两种结构,一种是没有分子内氢键的异构体1,另外一种是具有分子内氢键的异构体2。采用DFT方法〔4〕和Becke三参数杂化函数B3LYP〔5-6〕,结合全电子扩展基组6-31+G*和6-311++G**〔7-8〕,对两种异构体进行计算。计算工作是采用高级量子化学程序Guassian 09W〔9〕进行的。首先,在B3LYP/6-31+G*水平上,优化了planchol E的几何构型,并计算了谐性振动频率。由于发现没有虚频,表明所优化的结构,其能量已达到局域最小值。然后,在B3LYP/6-311++G**水平上,计算了H1NRM谱的化学位移。最后,采用含时密度泛函方法(TDDFT)〔10〕,在B3LYP/6-31+G* 理论水平上,求解前20个激发态。在求解过程中,电子旋转强度张量元由求得,其中为电偶极矩矢量为磁偶极矩矢量〔11〕。采用自编的小程序,拟合了具有洛伦兹线型特征的UV谱和ECD谱(20 nm半波宽),以及IR谱和VCD谱(30 cm-1半波宽,并采用0.962的频率校正因子)。
2 结果与讨论
2.1 分子结构和核磁共振谱(NRM)优化的分子结构见图1,planchol E具有四个手性碳中心,为RRRR结构。在图1(a)中,异构体1的两个六元环基本上是共面的(二面角小于3°),含氧六元环与中间含氧五元环之间二面角CCCC为157°和OCCO为75°;两个含氧五元环之间二面角CCCO为120°和OCCC为117°。分子骨架为扭曲的双螺旋结构。分子中间的C=O键为1.222 Å,C-OH中的C-O键为 1.353 Å,O-H 键为 0.971 Å。在图 1(b)中,异构体2有一个分子内氢键,导致比异构体1更大的扭曲,而且比异构体1更稳定(能量低了37.2 kJ/mol)。两个六元环是共面的,二面角约为2°。含氧六元环与中间含氧五元环之间二面角CCCC为87°和OCCO为160°。两个五元环之间二面角CCCO为96°和OCCC为138°。由于分子内氢键,C=O键被拉长了(1.241 Å),C-OH 中的 C-O 键缩短了 (1.340 Å),O-H 键拉长了(0.994 Å),H…O氢键为1.720 Å。
计算的H1NRM谱见图2。在图2(a)中,由于异构体1中没有分子内氢键,两个OH上的H化学位移相近,分别为11.2和11.4 ppm。实验上〔3〕,在氘代二甲亚砜(DMSO-d6)溶液中,测定的化学位移数值分别为11.1和11.7 ppm。可见,计算值与实验值相符。在图2(b)中,异构体2中的分子内氢键,使参与氢键的OH中的H具有强烈的去屏蔽作用,计算化学位移为19.5 ppm。没有参与分子内氢键的OH中的H的化学位移计算值为11.1 ppm。

图2 在B3LYP/6-311++G**水平上计算的核磁共振谱:异构体 1(a)和异构体 2(b)
作为一种气相单分子近似,计算结果表明:在气相中异构体2比异构体1稳定。但是,由于溶剂效应等因素影响,使实验测定的H1NRM谱与异构体1的计算结果更接近。我们推测,在测定NRM谱的实验条件下,planchol E主要以异构体1存在。
2.2 电子吸收光谱(UV)和电子圆二色谱(ECD)计算的UV谱、根据实验拟合的UV谱、计算的ECD谱(920 nm半波宽),见图3。
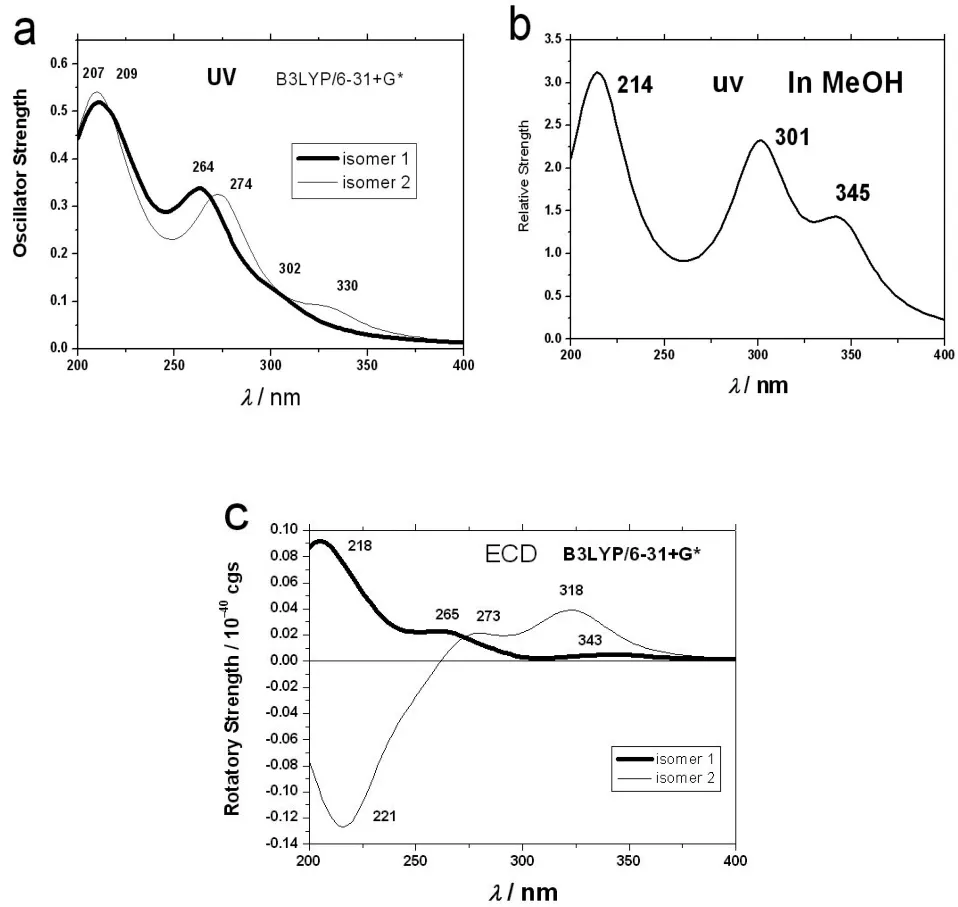
图3 计算的UV谱(a)、根据实验拟合的UV谱(b)、计算的ECD谱(c)(920 nm半波宽)
根据图3(a),计算的电子吸收光谱有三个吸收峰。异构体1的吸收波长分别为302、264和209 nm。异构体2的吸收波长分别为330、274和207 nm。根据图3(b)所示,在化合物1的甲醇溶液中〔3〕,测定的实验值分别为345、301和204 nm。虽然异构体2的计算值更接近在甲醇溶液中的实验值,但仅凭这一点,我们仍然无法把实验中的光谱归属到哪一个异构体。在甲醇溶液中,异构体2更可能稳定一些。在图3(c)中,我们发现两种异构体的ECD谱差异很大:异构体2在318 nm附近有强的正Cotton效应,而异构体2却很弱在343 nm处有弱的正Cotton效应;异构体2在221 nm处有强的负Cotton效应,而异构体1在218 nm处有强的正Cotton效应。
异构体1在218 nm处的强峰起源于S0→S11的电子跃迁,电子从HOMO-4跃迁到LUMO轨道上。可知,HOMO-4的电荷布居在整个分子骨架上(见图4),而LUMO的电荷主要布居在两个六元环上,因此,电子跃迁导致了电荷密度发生了从五元环到六元环的转移,产生了荷移峰。在343 nm处的峰起源于S0→S1的电子跃迁,是电子从HOMO-2跃迁到LUMO的结果,图4表明这两个分子轨道的电荷主要布居在苯环上,因此是电荷局域峰。

图4 异构体1对ECD谱有重要贡献较大的分子轨道电荷分布
异构体2在318 nm处的峰主要源于S0→S2的贡献,是电子从HOMO-2跃迁到LUMO的结果,可知,这个峰主要是苯环电荷局域峰(见图5)。在221 nm处的峰是S0→S8的电子跃迁结果(电子从HOMO-3跃迁到LUMO+3)。图5表明电荷密度从五元环转移到苯环,因此是一个荷移峰。

图5 异构体2对ECD谱有较大贡献的分子轨道电荷分布
此外,我们计算了比旋度,异构体1的比旋度是+116°,异构体2的比旋度是+247°,都比在甲醇中的实验值大(-26.0°)〔3〕。由于计算值和实验值符号相反,这一结果提示我们,我们计算的异构体可能是实验中的异构体的光学对映体,需要更多的证据。
2.3 红外振动光谱(IR)和振动圆二色谱(VCD)计算结果见图6。

图6 计算的红外振动吸收光谱(IR光谱(a)和振动圆二色谱(b))(半波宽30 cm-1)
图6(a)为计算的IR谱。在3 606 cm-1处吸收来源于O-H伸缩。两个异构体的IR谱在高频区差别很大。对异构体1,在2 952 cm-1处有较弱的吸收,由五元环上的CH2基团的C-H伸缩,在1 783 cm-1处的吸收为五元环上的C=O伸缩。苯环骨架伸缩导致了在1 601 cm-1处的吸收峰。1 078 cm-1处的吸收是两个五元环骨架伸缩结果。
对于异构体2,参与形成分子内氢键的羟基的O-H伸缩吸收峰出现在3 167 cm-1处,CH3中的C-H伸缩出现在3 016 cm-1处。1 786 cm-1处的吸收峰,是五元环上的C=O伸缩结果。在1 629 cm-1处的吸收,与苯环骨架伸缩有关。在1 139 cm-1处的吸收峰,来源于整个分子骨架伸缩贡献。
采用KBr做压片的planchol E样品,实验测定表明:在3 415 cm-1处有强吸收峰〔3〕。这个峰只能起源于参与分子内氢键的羟基的O-H伸缩。未校正的计算结果是3 292 cm-1,校正后是3 167 cm-1(见图1)。据此推测,在实验条件下,化合物主要以异构体2存在。我们把1 760 cm-1处的实验归属为C=O伸缩(计算值1 786 cm-1),1 658 cm-1处的吸收峰归属于苯环振动(计数值1 629 cm-1),1 170 cm-1处的吸收峰归属于整个分子骨架的振动(计数值1 139 cm-1)。
在图6(b)中,计算的两个异构体的VCD谱差别很大。异构体1在2 952 cm-1处有弱的负Cotton效应,为五元环上CH2的C-H伸缩结果。最强峰出现在1 218 cm-1处,是苯环与端点五元环骨架伸缩结果。
在异构体2的VCD谱中,在3 167 cm-1处有一个较强的负Cotton效应,是参与形成分子内氢键的羟基O-H伸缩结果。在1 210 cm-1处有一个强的负Cotton效应,是苯环与端点五元环伸缩的结果,在1 084 cm-1处有一个强的正Cotton效应,是两个五元环伸缩的结果。
3 结论
在本工作中,我们采用密度泛函方法,计算了化合物planchol E的两个异构体的核磁共振谱、电子吸收光谱、电子圆二色谱、红外振动光谱和振动圆二色谱,并进行了分子轨道和振动模式分析。计算结果与实验比较,推测在测定核磁共振谱的条件下,由于没有观察到氢键羟基H的化学位移,planchol E主要以没有分子内氢键的异构体1的形成存在;而在测定红外振动光谱的条件下,观察到氢键羟基的振动吸收峰,表明planchol E主要以具有分子内氢键的异构体2的形式存在。因此,planchol E是以分子内氢键的形式存在,会随实验环境的变化而变化。
此外,我们理论上预言了两种异构体的手性光谱(ECD和VCD谱),发现它们的手性光谱显著不同。在ECD谱中,异构体1在218 nm附近具有强的正Cotton效应,而异构体2在221 nm附近却出现强的负Cotton效应。在VCD谱中,异构体2在3167cm-1处出现较强的负Cotton效应,起源于分子内氢键的OH伸缩。
〔1〕Wang B,Ju J,He X F,et al.Three new terpenoids from Pinus yunnanensis〔J〕.Helv Chim Acta,2010,93(3):490-496.
〔2〕杨燕,杨茂发,杨再华,等.云南松松针的挥发性化学成分〔J〕.林业科学,2009,45(5):173-177.
〔3〕Lei T,Li Y,Li D,et al.A novel phenolic compound from Pinus yunnanensis〔J〕.J Asian Nat Prod Res,2011,13(5):425-429.
〔4〕Parr R G,Yang W.Density-functional theory of atoms and molecules〔M〕.Oxford:Oxford University Press,1989.
〔5〕Becke A D.Density-functional exchange-energy approximation with correct asymptotic behavior〔J〕.Phys Rev A,1988,38(6):3098-3100.
〔6〕Lee B,Yang W,Parr R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density〔J〕.Phys Rev B,1988,37(2):785-789.
〔7〕Petersson G A,Al-Laham M A.Open-shell systems and the total energies of the first-row atoms〔J〕.J Chem Phys,1991,94(9):6081-6090.
〔8〕Petersson G A,Bennett A,Tensfeldt T G,et al.The total energies of closed-shell atoms and hydrides of the firstrow elements〔J〕.J Chem Phys,1988,89(4):2193-2218.
〔9〕Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 09〔CP/DK〕.Wallingford CT:Gaussian,Inc.,2009.
〔10〕Bauernschmitt R,Häser M,Treutler O,et al.Calculation of excitation energies within time-dependent density theory using auxiliary basis set expansions〔J〕.Chem Phys Lett,1997,264(6):573-578.
〔11〕Moscowitz A.Theoretical aspects of optical activity.Part one:small molecules〔J〕.Adv Chem Phys,1962(4):67-112.
Density Functional Study of Chiral Structures and Spectral Properties of the Phenolic Compound Planchol E from Pinecones of Pinus yunnanensis
YANG Ying1,LIU Guangming1,LEI Ting1,MANG Chaoyong1,2*
(1.College of Pharmacy and Chemistry,Dali University,Dali,Yunnan 671000,China;2.Institute of Pharmaceutical Science,Dali University,Dali,Yunnan 671000,China)
The research on the relationship between stereo-chemical structures and chiral spectra is a fundamental job for the applications of chiral compounds in chiral materials and chiral drugs.The DFT computations of a chiral phenolic compound called as planchol E show that the experimental H1NRM spectra display the feature of isomer 1 without intramolecular H-bond while the measured IR spectra originate from isomer 2 with intramolecular H-bond.Due to the intramolecular H-bond in isomer 2,there are many differences in both the chiral spectra as ECD and VCD and the achiral spectra as NRM and IR.
plancholE;densityfunctionaltheoreticalmethod;electroniccirculardichroism;vibrationalcirculardichroism;hydrogen bond
O64
A
1672-2345(2013)04-0040-04
国家自然科学基金资助项目(81260632)
2013-01-01
2013-02-20
杨颖,硕士研究生,主要从事药物化学和物理化学研究.
(责任编辑 袁 霞)
10.3969/j.issn.1672-2345.2013.04.011
*通信作者:莽朝永,教授,博士.
