X-连锁无丙种球蛋白血症BTK基因新发突变1例报告
2013-04-09陶英博陈慧珊曾华松
陶英博,陈慧珊,曾华松
(广州医学院附属广州市妇女儿童医疗中心 广州市儿童医院过敏免疫风湿科,广州 510120)
X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia, XLA)又称Bruton无丙种球蛋白血症,是最早发现的人类原发性免疫缺陷病(primary immunodeficiency disease, PID),由Bruton于1952年首次报道,为PID发病率最高的类型之一。现报道1例基因确诊病例,以提高儿科医师对该类疾病的认识。
临床资料
患儿男,5岁,汉族,因反复发热伴咳嗽4 d就诊。患儿自6月龄开始,反复出现咳嗽、咯痰、发热,多次诊断为“支气管肺炎,肺部感染”,抗感染治疗后症状好转,但易反复。此次患儿无明显诱因出现发热,夜间明显,体温最高可达40℃,口服退热药后症状缓解但反复发生,伴寒战、轻度咳嗽,无抽搐及皮疹、无痰、无流涕及呕吐。
既往史及家族史:第1胎、第1产,足月剖宫产,生于医院,出生体重2.55 kg,无窒息史。患儿为α-地中海贫血患者,平素体制差,易“感冒”发热,无外伤及手术史。父母体健,非近亲结婚,其父为α-地中海贫血患者,家族中无明显遗传病及传染病史。
血常规检查:白细胞计数10.2×109L,淋巴细胞绝对值3.18×109L,中性粒细胞百分比0.61,淋巴细胞百分比0.31,嗜酸粒细胞百分比0.01,红细胞计数6.09×1012L,血红蛋白119 gL,红细胞压积37.1,血小板计数585×109L。体液免疫检查:IgG<0.33 gL,IgA<0.07 gL,IgM<0.04 gL,补体C3 1.34 gL,补体C4 0.19 gL,IgE<5 Uml。流式细胞仪检查:CD3+CD45+比例87%,CD3+T细胞绝对计数3498个μl,CD3+4+T细胞绝对计数1491个μl,CD3+4+T细胞比例37%,CD3+8+T细胞绝对计数1598个μl,CD3+8+T细胞比例40%,CD19+B细胞绝对计数30个μl,CD19+B细胞比例1%,CD16+56+自然杀伤细胞绝对计数340个μl,CD16+ 56+自然杀伤细胞比例9%。肝功能、尿、粪常规检查未见异常。病毒性乙型肝炎各项抗体均为阴性。X线胸片示支气管肺炎。
分析患儿病情,患儿生后6个月开始出现体质差,表现为易“感冒”、发热、肺部感染,对症及抗感染治疗后好转,之后易反复。追问患儿家族史,家族中无儿童早期感染病死情况,但临床表现及实验室检查诊断PID可能性大,进一步行基因突变检测以明确诊断。
取患儿新鲜全血200 μl,根据BTK基因序列设计引物,采用聚合酶链反应扩增,产物直接测序。DNA同源性分析使用NCB I BLAST程序,参考BTK基因序列(U78027,X58957)。结果发现,患儿X染色体BTK基因第14个外显子存在置换突变,由A突变为T(图1),使编码蛋白质由精氨酸变为终止密码子,从而终止转录。
讨 论
XLA患者以反复细菌感染为特征,血清中各类免疫球蛋白明显降低或缺乏,对抗原刺激不能产生抗体应答,血循环中B淋巴细胞减少,淋巴结及淋巴组织缺乏生发中心和淋巴滤泡,骨髓中无浆细胞,但前B淋巴细胞数量正常,T淋巴细胞数量及功能正常。患儿往往于6~8月龄起病,反复出现肺炎双球菌、链球菌等化脓性感染,导致咽炎、肺炎、脑膜炎、败血症等,13患儿同时伴发幼年型类风湿性关节炎,约半数患儿于10岁前死亡。
XLA临床诊断依据:(1)男性;(2)反复较严重细菌感染(呼吸道、胃肠道、皮肤及其他深部感染),抗生素治疗效果不佳;(3)伴或不伴有自身免疫性疾病;(4)伴或不伴有母系家族中类似疾病表现的男性患者[1]。最主要的辅助诊断依据为血IgG<2 gL及外周血成熟B淋巴细胞缺失或明显降低(<1%)[2]。XLA的临床表现以呼吸道感染为主。由于XLA患儿存在反复感染,所以普遍存在营养不良和生长发育迟缓。扁桃体和浅表淋巴结发育不良是该病患儿的特征性体征。尽管存在反复细菌感染,局部淋巴组织亦没有增生现象。这也是临床最容易观察到的现象。
BTK基因定位于X染色体q21.3-22,基因全长37.5 kb,含有19个外显子,除第1外显子外,其余18个用以编码BTK蛋白的659个氨基酸,相对分子质量为76 000[3]。BTK蛋白是非受体型蛋白酪氨酸激酶Tec家族的一员,能催化多种底物蛋白质酪氨酸残基磷酸化,在B淋巴细胞发育的信号转导中起重要作用。Tec家族成员还包括TEC、ITK、TSK、EMT和BMN。TEC家族蛋白酪氨酸激酶包括5个不同的结构区段(图2),从N末端起为Pleckstrinhomology(PH)段,约有120个氨基酸,Techomology(TH)段约有60~80个氨基酸,Srchomolgy3(SH3)段约有60个氨基酸,SH2段约100个氨基酸,激酶催化段约280个氨基酸[4]。
BTK基因突变不仅存在于19个外显子,还涉及内含子和启动区域。其中,发生频率最高的是错义突变,其次是无义突变和拼接位点突变。错义突变主要发生在三联体密码子的前两位。值得注意的是,并非所有的XLA患儿都存在BTK基因突变。约10%~20%的B淋巴细胞免疫缺陷患儿(男女均可发病)的BTK基因并无异常,提示XLA可能存在其他基因异常[5]。
替代治疗和造血干细胞移植是目前PID治疗的两大重要手段,基因治疗仍在研究及开发之中。此前,我国儿科医师在合理治疗方案选择和个体化方面存在明显不足,以致出现患儿过早夭折、生存质量低下和过度治疗等问题。常规治疗方法是在出现严重、致命性感染前行免疫重建,早期诊断和骨髓移植明显提高了长期生存率[6]。但由于移植配型困难,目前对于XLA的基因治疗寄予厚望。有研究表明,及早给予免疫球蛋白替代治疗,大多数患儿预后较好[7],但应用丙种球蛋白终生替代治疗费用昂贵,长期应用依从性差。
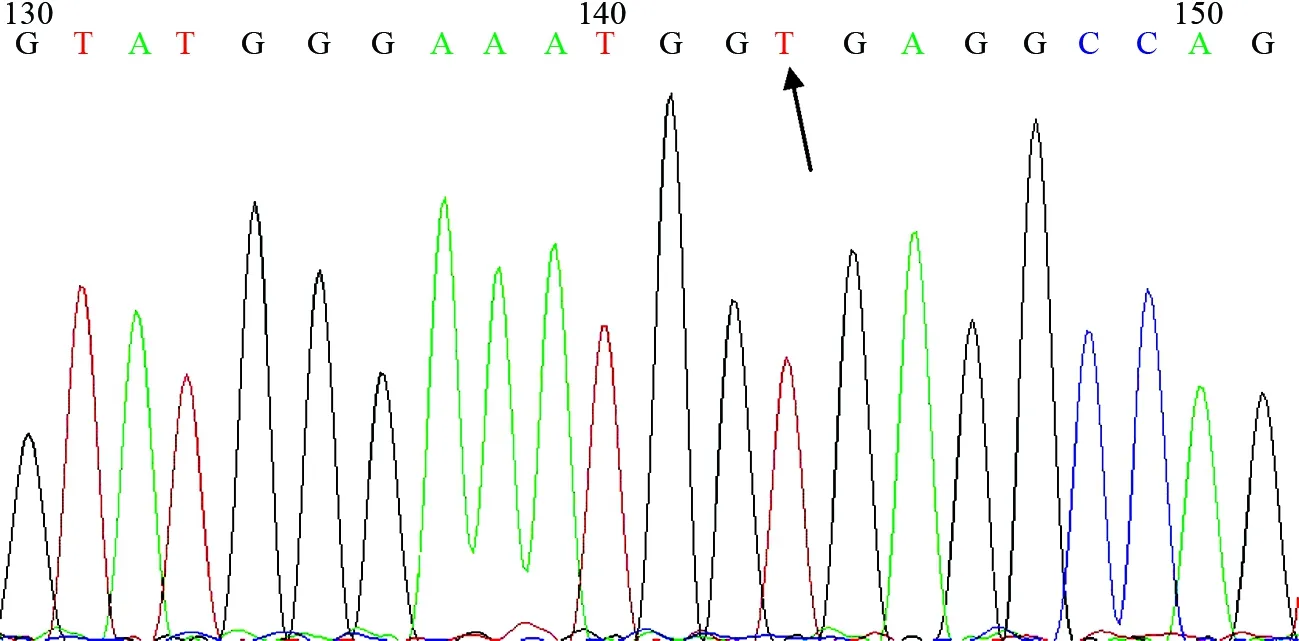
图1 BTK基因第14个外显子测序图箭头示碱基A突变为T

图2BTK蛋白组成示意图
从N端开始5个区域:Pleckstrinhomology(PH)、Techomology(TH)、Srchomolgy3(SH3)、SH2和激酶区
图上方数字:各段氨基酸长度;图下方数字:外显子
本例患儿BTK基因DNA测序发现单核苷酸置换突变,其第14外显子碱基由A置换为T。由于多种原因的限制,未能对患儿父母进行基因检测,遂未了解其父母携带情况,未能进行家系调查,是本报告的不足之处。据患儿父母口述,其双方家族均无遗传病史,若基因测序后证实双方父母及家系确无携带基因,则本患儿可能为家族中首例基因突变。目前,已开展了产前基因突变检测,可以针对有家族史患者进行筛查[8]。
[1]杨锡强. 原发性免疫缺陷病的协作网和登记工作[J]. 中华儿科杂志, 1999, 37:328-329.
[2]陈同辛, 王玺. 原发性免疫缺陷病诊断标准[J]. 实用儿科临床杂志, 2006, 21:573-576.
[3]王剑利, 耿松海. BTK基因与XLA[J]. 国外医学 遗传学分册, 2000, 23:263-266.
[4]Vihinen M, Kwan SP, Lester T, et al. Mutation of the human BTK gene coding for Bruton tyrosine in X-linked agammaglobulinemia[J]. Human mutation, 1999,13:280-285.
[5]Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patients with defects in early B-cell development[J]. Immunol Rev, 2005,203:16-234.
[6]Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution[J]. Annu Rev Immunol, 2004, 22:25-655.
[7]Granados EL, Porpiglia AS, Hogan MB, et al. Clinical and molecular analysis of patients with defects inμ heavy chain gene[J]. J Clin Invest, 2002,110:1029-1035.
[8]Li L, Zhou Y, Wang J, et al. Prenatal diagnosis and carrier detection for Athabascan severe combined immunodeficiency disease[J]. Prenat Diagn, 2002, 22:763-768.
