聚右旋丙交酯和聚左旋丙交酯乙交酯的制备及其共混物的结晶行为*
2013-03-26黄冬玲陈栋梁熊成东熊左春
黄冬玲,董 军,陈 龙,陈栋梁,熊成东,熊左春
(1.中国科学院成都有机化学研究所,四川成都 610041;2.中国科学院大学,北京 100049)
聚乳酸及其共聚物因原料来源广泛,且具有良好生物相容性、可降解性而获得了广泛而深入的研究和应用[1~4],如医用植入材料、组织工程支架、药物控释载体等方面。为了进一步改善聚乳酸相关性能,目前主要有化学改性和物理改性两种方法,前者有如和己内酯无规共聚改善其柔韧性,和乙交酯无规共聚调节其降解速度,和聚乙二醇的嵌段共聚改善其亲水性能等;后者包括通过添加如无机纳米粒子改善其力学性能和生物学性能,改变加工条件调节其聚集态结构等。
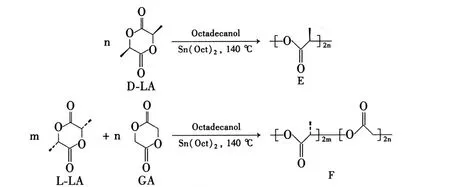
Scheme 1
针对聚乳酸及其共聚物的耐热性不足问题,目前主要采用提高其结晶度,与耐热型聚合物的共混技术、辐射交联技术、纳米复合技术等来提高其耐热性,而采用立构复合技术是近年来发展耐热型的方向[5~8]。聚左旋乳酸结构单元和聚右旋乳酸结构单元在一定条件下可以通过分子链间较强的范德华力作用形成一种立构复合物,其熔点比各自均聚物的熔点高30℃以上,有效地提高了聚乳酸及其共聚物的耐热性。
本文以辛酸亚锡为催化剂,通过本体熔融开环聚合法合成了聚右旋丙交酯(E)和聚左旋丙交酯乙交酯(F),其结构经1H NMR和IR表征。并研究了E和F共混物(M)的结晶行为。
1 实验部分
1.1 仪器与试剂
BRUKER AVANCE 300 NMR型核磁共振仪(CDC13为溶剂,TMS为内标);NICOLET-iS10型红外光谱仪(KBr压片);TA Q20型差示扫描量热仪(升温速率10℃·min-1);乌氏粘度计(氯仿为溶剂,测定温度25℃)。
右旋丙交酯(D-LA),左旋丙交酯(L-LA)和乙交酯(GA),自制;辛酸亚锡,Sigma公司;其余所用试剂均为分析纯。
1.2 合成
(1)E 的合成[9]
在反应瓶中依次加入 D-LA 72.0 g(500 mmol)、辛酸亚锡 0.10 g(0.25 mmol)和十八醇1.35 g(5 mmol),抽真空(<10-3Pa),搅拌下于140℃反应24 h。加入氯仿溶解,用过量乙醇沉析,过滤,滤饼经室温真空干燥至恒重得E0.6(特性粘度为 0.60 dL·g-1)备用。
改变十八醇用量,按类似方法合成 En[E2.0(1.0 mmol)和 E4.0(0.25 mmol)]。
(2)F 的合成[10]
在反应瓶中依次加入 L-LA 68.8 g(478 mmol),GA 2.6 g(22 mmol),辛酸亚锡 0.10 g(0.25 mmol)和十八醇68 mg(0.25 mmol),按1.2(1)方法合成 F4.0。
1.3 M的制备(以为例)
在反应瓶中加入 E0.60.04 g 和 F4.00.96 g,氯仿10 mL,搅拌使其溶解;于室温自然挥干,于95℃真空干燥2 h得{w=m(E)/[m(E)+m(F)]×100%=4%}。
分别用 E2.0和 E4.0代替 E0.6,按类似方法制得和(w=4%,9%,20%,40%,49%,60%,75%)。
2 结果与讨论
2.1 表征

图 1 En和 F4.0的 IR 谱图Figure 1 IR spectra of Enand F4.0
E 和F的1H NMR 分析表明,5.1~5.2和1.55~1.60为E中D-LA结构单元的次甲基和甲基的吸收峰;5.1 ~5.2,4.6 ~4.9 和1.55 ~1.60 分别为F中L-LA结构单元的次甲基,GA结构单元的亚甲基和L-LA结构单元的甲基的吸收峰。
图1为En和F4.0的 IR谱图。由图1可见,E0.6,E2.0和 E4.0均为 D-LA 的均聚物,官能团结构没有差别,仅在结构单元数量上不同。En的IR谱图无明显差异,均在2 990 cm-1和2 940 cm-1处出现饱和碳氢伸缩吸收峰,1 760 cm-1处的强吸收峰和1 050~1 250 cm-1处的强吸收双峰为羧酸酯特征峰,1 460 cm-1处和 1 350~1 400 cm-1附近为甲基C-H弯曲振动吸收峰。F4.0的IR谱图与E相似。

图2 M的DSC曲线Figure 2 DSC curves of M
2.2 M的结晶行为分析
图2 为M的DSC曲线。由图2可见,随着w(E0.6)的增加,在 160 ℃ 附近的 F4.0晶体吸热峰逐渐减弱至消失,而170℃附近E0.6的晶体吸热峰出现并逐渐增强,同时210℃附近的立构复合物(SC)[8]吸热峰强度先逐渐增加,在出现极大值后强度开始降低。由图2还可以看出,除SC吸热峰极大值点出现在和外,其余 M 中随w(E)增加的趋势同图 E0.6。对比,和的一次升温曲线,可以看到随E分子量的增加,M熔融吸热峰峰形发生明显变化;在中,E0.6晶体吸热峰强度要显著小于SC吸热峰强度,在中,E2.0和 F4.0混合峰强度和SC吸热峰强度相当;而在中SC吸热峰变小,明显弱于 E4.0和 F4.0混合峰强度,这种分子量的影响在二次升温曲线中更为明显。
由于一次升温时M经过95℃退火处理[10],使其分子链得到充分松弛,结晶也相对完善,其熔融吸热峰强度可以反映共混体系的最大结晶能力;二次升温曲线则反映M在实验条件下的实际结晶速率。从图2可以看出,无论是 160 ℃附近的F4.0晶体熔融吸热峰,还是210℃附近的SC熔融吸热峰都明显减弱,说明在二次升温的条件下,由于结晶速度的限制未能充分结晶,其余 M情况类似。
图3为根据M的一次升温曲线获得的熔融焓(ΔHm)和共混物比例之间的关系。ΔHm1为150℃~190℃间出现的所有E和F的熔融吸热峰对应的吸收焓值,ΔHm2为SC吸收焓值。由图3可见,w(E0.6)为 4% 时,ΔHm1大于 ΔHm2,说明此时M 熔融焓主要是 F4.0熔融所贡献;随着w(E0.6)的增加(9% ~60%),ΔHm1减小的同时 ΔHm2增大并超过前者,说明SC含量逐渐增加,并构成M的主要结晶相;w(E0.6)进一步增大(75%),由于 F4.0含量减少,导致ΔHm2减小而相应的ΔHm1增加,而此时的ΔHm1变为主要由E0.6熔融贡献,按SC形成原理[8,11]推理,ΔHm2极大值应该出现在中,但实际出现在中,这是由于高分子量的 F4.0分子链运动受到限制,而稍过量的低分子量E0.6在和其形成SC的同时起到润滑分子链的作用。由图3还可以看出,和的 ΔHm1和 ΔHm2的总体变化趋势与相似,均随w增加,在w=49%附近出现ΔHm2极大值;但随着E分子量的增加(E2.0和 E4.0),只有w=49% 附近 ΔHm2大于ΔHm1,说明其形成SC的能力开始减弱;而在中,ΔHm2远小于 ΔHm1,说明 E4.0和 F4.0仅能形成少量SC,w(E4.0)=9%时,DSC检测未发现 SC熔融峰。
从图3也能看出,小分子量的E0.6加入,可以促进M的结晶,其熔融焓(ΔHm1和 ΔHm2)达44 J·g-1以上,且SC的形成能力较强;而较高分子量的 E2.0和 E4.0与 F4.0共混时则阻碍了 M 结晶,熔融焓最大值分别为34 J·g-1和28 J·g-1。由于SC的形成是D-乳酸单元和L-乳酸单元按等摩尔比配对形成的,所以SC的形成和E(或F)自身结晶存在互相制约关系,SC含量增加时E(或F)自身结晶含量减少。
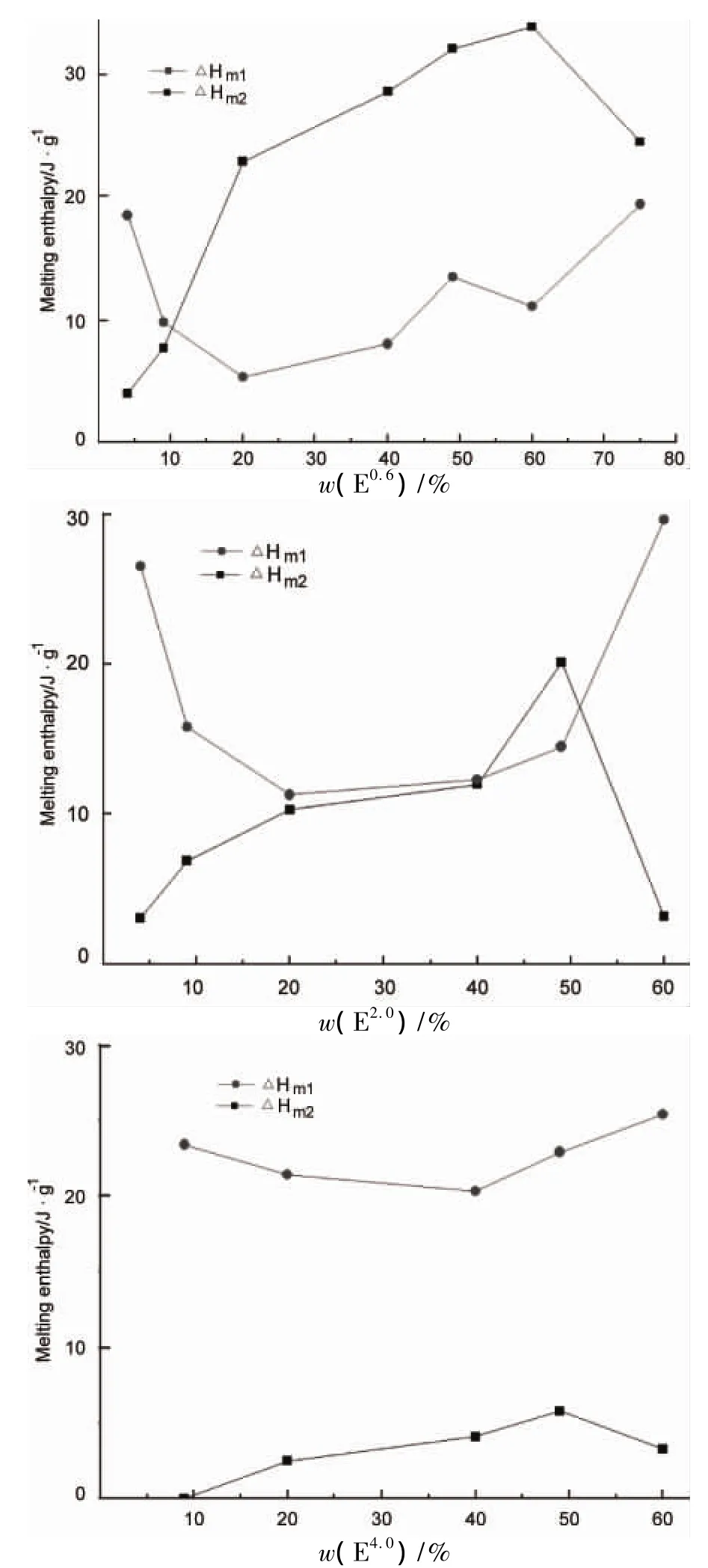
图3 M的熔融焓Figure 3 Melting enthalpy of M
3 结论
通过本体熔融开环聚合制备了系列右旋丙交酯聚合物(E)和左旋丙交酯乙交酯共聚物(F),并将二者以不同比例进行共混,共混物M的结晶行为研究结果表明,通过调节E的分子量和共混比例,可以调节M的结晶能力和结晶形态,能够提高聚乳酸类材料的使用温度,从而满足部分实际应用的要求。
[1]Auras R,Lim L,Selke S,et al..Poly(lactic acid)Synthesis,Structures,Properties,Processing,and Application[M].America:John Wiley & Sons,Inc,2009:445-484.
[2]Sawai D,Takahashi K,Sasashige A,et al.Preparation of oriented β-form poly(L-lactic acid)by solidstate coextrusion effect of extrusion variables[J].Macromolecules,2003,36(10):3601 -3605.
[3]Nair L S,Laurencin C T.Biodegradable polymers as biomaterials[J].Progress in Polymer Science,2007,32:762-798.
[4]Abraham J,Neeraj K,Aviva E.Biodegradable polymers in clinical use and clinical development[M].Singapore:John Wiley & Sons,Inc,2011:319 -350.
[5]李菁,陈大凯,任杰.聚乳酸立构复合物的研究最新进展与应用展望[J].高分子通报,2011,1:33-38.
[6]Tsuji H,Fukui I.Enhanced thermal stability of poly(lactide)s in the melt by enantiomeric polymer blending[J].Polymer,2003,44:2891 -2896.
[7]Purnama P,Jung Y,Kim S H.Stereocomplexation of poly(l-lactide)and random copolymer poly(d-lactideco-ε-caprolactone)to enhance melt stability[J].Macromolecules,2012,45(9):4012 -4014.
[8]Tsuji H.Poly(lactide)stereocomplexes:Formation,structure,properties,degradation,and applications[J].Macromolecular Bioscience,2005,5(7):569 -595.
[9]熊左春,陈栋梁,李庆,等.不同旋光度PLGA的制备及其结晶性能研究[J].合成化学,2009,17(3):292-295.
[10]Park S D,Todo M,Arakawa K,et al.Effect of crystallinity and loading-rate on modeⅠfracture behavior of poly(lacticacid)[J].Polymer,2006,47:1357-1363.
[11]Wang X H,Prud'homme R E.Differences between stereocomplex spherulites obtained in equimolar and non-equimolar poly(L-lactide)/poly(D-lactide)blends[J].Macromolecular chemistry and physics,2011,212(7):691-698.
