(2S,3S)-1,2-环氧基-3-叔丁氧酰胺基-4-苯丁烷的合成*
2013-03-26谌志华马红梅虞心红
谌志华,徐 辉,庄 璐,盖 涛,马红梅,虞心红
(1.上海奥奇医药科技有限公司,上海 200032;2.华东理工大学药学院上海市新药设计重点实验室,上海 200237)
HIV蛋白酶抑制剂作为抗艾滋病的一类药物,其作用机制是通过抑制蛋白酶对gag和gagpol蛋白的水解过程,扰乱病毒的成熟过程,使感染细胞释放出不成熟和有缺陷的病毒颗粒,从而抑制病毒的有效复制[1]。目前已上市的药物有沙奎那韦、安普那韦、阿扎那韦、福沙那韦、替拉那韦和地瑞那韦等。临床研究表明,单独使用此类药物4~12周,体内的病毒会降低2~3个数量级;与NRTI联合使用,60% ~95%患者体内病毒会降至血液中检测不出[2]。HIV-1蛋白酶抑制剂是高效抗逆转录病毒疗法(HAART)的重要组成部分,自HAART应用于临床以来,HIV感染者死亡率明显降低。但该类药物有脂肪重新分布、血症代谢异常及肝毒性血糖升高等副作用和耐药性(尤其是交叉耐药性)[3]。
(2S,3S)-1,2-环氧基-3-叔丁氧酰胺基-4-苯丁烷(1)是合成HIV-1蛋白酶抑制剂,如沙奎那韦、安普那韦、福沙那韦、地瑞那韦等的关键中间体,其常用合成方法一般会用到有毒易爆炸的重氮甲烷[4],或是使用价格昂贵的锂试剂反应[5,6],这两种方法对设备及操作要求较高,并且合成过程安全性较差。
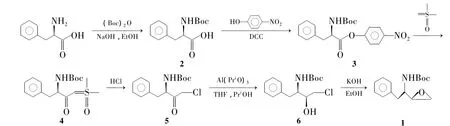
Scheme 1
文献[7]以N-叔丁氧羰基-(S)-苯丙氨酸对硝基苯酚酯(3)为起始原料,经与硫叶立德反应制得(S)-1-氯-3-叔丁氧酰胺基-4-苯基-2-丁酮(5);5经硼氢化钠还原、氢氧化钾环合等反应得到1。该路线避免了使用重氮甲烷和锂试剂,操作安全性大大提升。
本文参考文献[8,9]方法合成了 1(Scheme 1),并对其工艺进行了改进和优化:以(S)-苯丙氨酸为起始原料,用Boc2O保护氨基制得N-叔丁氧羰基-(S)-苯丙氨酸(2)时,后处理未按文献[8](收率78% ~87%)方法进行结晶,而是直接投入下步反应合成3,避免了产品损耗,两步收率达到82%,较文献提高17% ~23.5%;合成5时,使用氯化氢的二氧六环溶液替代氯化锂和甲磺酸作为氯源,降低了成本;用Meerwein-Poondrf-Verley还原替代硼氢化钠还原制备(2S,3S)-1-氯-3-叔丁氧酰胺基-4-苯基-2-丁醇(6),收率由50%[7]提高至83.5%。
该工艺总收率约45%,具有原料价廉易得,反应条件温和,操作简单,不需柱层析纯化等优点,适合工业化生产。
1 实验部分
1.1 仪器与试剂
WRS-1B型熔点仪(温度未校正);BRUKER AVANCE 400型核磁共振仪(CDCl3为溶剂,TMS为内标);Micromass GCT型质谱仪。
(S)-苯丙氨酸和硫叶立德,工业品;其余所用试剂均为分析纯。
1.2 合成
(1)N-叔丁氧羰基-(S)-苯丙氨酸(2)[8]的合成
在反应瓶中加入NaOH 4.45 g(110 mmoL)的水(100 mL)溶液和苯丙氨酸 16.50 g(100 mmoL),搅拌使其溶解;加入乙醇15 mL,冰浴冷却,滴加(Boc)2O 22.00 g(101 mmol)(10 min,反应液中有气泡冒出);于室温反应4.5 h。减压蒸除乙醇,用1 mol·L-1盐酸调至 pH 1 ~2,用乙酸乙酯(3×100 mL)萃取,合并有机层,用无水硫酸钠干燥,减压浓缩得浅黄色黏稠液体2 26.60 g(78% ~87%[8]),直接进行下步反应。
(2)N-叔丁氧羰基-(S)-苯丙氨酸对硝基苯酚酯(3)的合成
在反应瓶中加入2 23 g(86.69 mmoL)的乙酸乙酯(300 mL)溶液,对硝基苯酚12.66 g(91.03 mmoL)和二环己基碳酰亚胺(DCC)18.78 g,搅拌下于20℃反应4 h。抽滤,滤饼用乙酸乙酯(3×45 mL)洗涤,合并滤液与洗液,依次用水(3×50 mL)和饱和NaHCO3溶液(3×50 mL)洗涤;合并水层,用乙酸乙酯(2×150 mL)萃取,合并有机层,用饱和食盐水(3×100 mL)洗涤,无水硫酸钠干燥,减压浓缩后用乙醇重结晶得黄色固体3 27.36 g,收率 82%(两步),m.p.128.6 ℃ ~128.7 ℃(75%,117 ℃ ~120 ℃[9]);1H NMR δ:1.47(s,9H),3.30 ~ 3.19(m,2H),4.82(dd,J=13.80 Hz,6.68 Hz,1H),5.11(d,J=7.61 Hz,1H),7.16(d,J=9.08 Hz,2H),7.25 ~7.43(m,5H),8.26(d,J=9.01 Hz,2H)。
(3)二甲基硫氧-(S)-2-氧代-3-叔丁氧酰胺基-4-苯丁基叶立德(4)的合成
将三甲基碘化亚砜16.50 g(75 mmoL)和叔丁醇钾8.42 g(75 mmoL)悬浮于 THF(75 mL)中,搅拌下回流反应1 h。冷却至室温,加入3 9.65 g(25 mmoL)的 THF(75 mL)溶液,反应 2.5 h(TLC跟踪)。加水25 mL,搅拌15 min;过滤,滤饼用乙酸乙酯(3×15 mL)洗涤,合并滤液与洗液,减压浓缩至约20 mL,加水50 mL,用乙酸乙酯(2×50 mL)萃取,合并有机层,依次用水(30 mL),饱和食盐水(2×30 mL)洗涤,无水硫酸钠干燥,浓缩得淡黄色固体4 8.50 g(直接进行下步反应),m.p.112.6 ℃ ~117.0 ℃(90% ~91%,163 ℃[7]);1H NMR δ:1.43(s,9H),2.97 ~3.07(m,1H),3.27(s,1H),3.37(s,1H),4.21 ~4.42(m,2H),5.26(d,J=8.16 Hz,1H),7.22 ~7.29(m,5H);MS-ESIm/z:340{[M+H]+},362{[M+Na]+},378{[M+K]+}。
(4)5的合成
在反应瓶中加入 4 8.00 g(23.57 mmoL)的THF(100 mL)溶液,盐酸3 mL和二氧六环6 mL,搅拌下回流反应2 h。冷却至室温,加入乙酸乙酯50 mL和正己烷100 mL,依次用水(2×100 mL),饱和NaHCO3溶液(50 mL),饱和食盐水(50 mL)洗涤,无水硫酸钠干燥,减压浓缩得粗品,用正己烷重结晶得类白色固体5 5.62 g,收率80.0%,m.p.103.8 ℃ ~ 104.4 ℃ (收率 81%,103 ℃[7]);1H NMR δ:1.43(s,9H),2.98 ~ 3.13(m,2H),4.00(d,J=16.20 Hz,1H),4.19(d,J=16.20 Hz,1H),4.69(m,1H),5.05(s,1H),7.19 ~7.37(m,5H);MS-ESIm/z:320{[M+Na]+},336{[M+K]+}。
(5)6的合成
在反应瓶中加入5 2.98 g(10 mmoL),异丙醇铝1.02 g(5 mmoL),异丙醇20 mL 和 THF 20 mL,搅拌使其完全溶解;于40℃反应5 h。减压浓缩至干,残余物加入二氯甲烷(50 mL)和水(5 mL),滴加1 mol·L-1盐酸至pH 2~3;分液,水层用二氯甲烷(20 mL)萃取,合并有机层,依次用水(10 mL),5%碳酸氢钠溶液(10 mL)洗涤,用饱和食盐水(4×10 mL)洗至pH 7,无水硫酸钠干燥,减压浓缩得淡黄色固体,用乙酸乙酯重结晶得白色固体6 2.50 g,收 率 83.5%,m.p.169.2 ℃ ~ 171.4 ℃(50%[7]);1H NMR(DMSO-d6)δ:1.27(s,1H),2.57(dd,J=13.76 Hz,9.71 Hz,1H),3.03(d,J=13.98,1H),3.45 ~ 3.69(m,4H),5.45(s,1H),6.72(d,J=8.62 Hz,1H),7.13 ~7.27(m,5H);MS-ESIm/z:322{[M+Na]+},338{[M+K]+}。
(6)1的合成
将6 1.32 g(5 mmoL)悬浮于乙醇20 mL中,冰浴冷却下滴加KOH 0.28 g(5 mmoL)的乙醇(5 mL)溶液,滴毕,于室温反应2 h;滴加冰醋酸至pH 4,减压浓缩得白色固体,用乙酸乙酯(50 mL)溶解,用饱和NaHCO3溶液(2×5 mL)洗涤,无水硫酸钠干燥,减压浓缩得白色固体,用正己烷重结晶得无色针状晶体 1 1.08 g,收率 82.0%,m.p.127.2℃ ~127.5 ℃,[α]28D-8.1°(c1,MeOH)[125 ℃,[α]25D- 8.3°(c1,MeOH)[6]];1H NMR δ:1.40(s,9H),2.78 ~3.02(m,5H),3.58 ~3.85(m,1H),4.52(s,1H),7.24 ~7.34(m,5H);MS-ESIm/z:286{[M+Na]+},302{[M+K]+}。
[1]Shetty B V,Kosa M B,Webber S,et al.Preclinical pharmacokinetics and distribution to tissue of AG1343,an inhibitor of human immunodeficiency virus type 1 protease[J].J Antimicrob Angents Chemother,1996,40(1):110-114.
[2]唐红梅.抗艾滋病复制周期药物研究的新进展[J].广东医学,2005,26(9):1286 -1288.
[3]Clercq E.New approaches toward anti HIV chemotherapy[J].J Med Chem,2005,48(5):1297 -1313.
[4]Joel C B,Eric G,Masud A,et al.Aminodiol HIV protease inhibitors.Design,synthesis and preliminary SAR[J].J Med Chem,1994,37(12):1758 -1768.
[5]Daniel P G,Gary A D,Heintz R M,et al.Discovery of a novel class of potent HIV-1 protease inhibitors containing the(R)-(hydroxyethyl)urea isostere[J].J Med Chem,1993,36(2):288 -291.
[6]Jose B,Beatriz B,Jose M C.Highly diastereoselective synthesis of threo and arythro aminoalkyl epoxides from αamino acids[J].J Org Chem,1995,60(21):6696 -6699.
[7]Wang Dengjin,Schwinden Mark D,Nugent William A,et al.One-carbon chain extension of esters to αchloroketones:A safer route without diazomethane[J].J Org Chem,2004,69(5):1629 -1633.
[8]Oskar K,Walter E K,Gert V L,et al.Tert-butoxycarbonylation of amino acids and their derivatives:N-tert-butoxycarbonyl-1-phenylalanine[J].Org Synth,1985,1(63):160 -166.
[9]Volkmann O B.Catalysis of an ester hydrolysis applying molecularly imprinted polymer shells based on an immobilised chiral template[J].Reactive and Functional Polymers,2006,66:1725 -1733.
