慢病毒Tet-On系统调控Notch1-EGFP融合蛋白在PC12细胞中表达的研究*
2013-03-19刘永敏黄春田韩雪飞鄢文海
刘永敏,段 萍,黄春田,李 博,韩雪飞,许 燕,鄢文海,邢 莹,4△
(1.郑州大学干细胞研究中心,2.病理生理学教研室,3.生理学教研室,河南郑州450001;4.新乡医学院生理学教研室,河南新乡453003)
Notch信号是一个在进化过程中高度保守的信号通路,Notch基因在许多组织细胞中都有表达,包括中枢神经系统、胰腺、造血细胞等,它编码一种跨膜受体蛋白,参与调节各种细胞分化及发育的信号转导途径[1-3]。Notch信号通路的失调与多种神经系统肿瘤形成密切相关。
在体内Notch信号通路始终处于一个动态调控过程中,因此建立一个可调控的研究体系有利于研究Notch信号通路在不同时间点的作用差异。在可调控的真核生物基因表达系统中,四环素调控系统被认为是目前最为理想的选择,该系统具有严密性、特异性、高效性、低毒性等优点,被广泛地应用到生物医学研究中。
在本研究中我们构建Tet-on调控的携带增强型绿色荧光蛋白(EGFP)的人源性 Notch1胞内段(Notch1 intracellular domain,NICD)融合蛋白的慢病毒真核表达载体,并探讨四环素基因表达系统调控Notch1-EGFP在大鼠嗜铬细胞瘤细胞系(PC12细胞)细胞中表达的时间和浓度的量效关系,为研究Notch信号通路的作用提供一条更灵活更科学的途径。
1 材料与方法
1.1 材料
1.1.1 人体胎盘组织 正常人胎盘组织取自河南省人民医院,根据国务院《医疗机构管理条例》规定,患者签写知情同意书,符合伦理学标准。
1.1.2 试剂 大肠杆菌 JM109、pEGFP-C1质粒、pcDNA 3.1(+)载体、PC12细胞为本实验室常规保存、pLVX-Tight-Puro载体,pLVX-Tet-On Advanced及Lenti-XTMHTX Packaging System载体购自ClonTech公司,总RNA提取试剂盒(Trizol)购自北京天根公司,cDNA第一链试剂盒购自formatas公司,限制性内切酶 BamH I、EcoR I、Nhe I、Xba I、T4连接酶、DNA Marker(1kb,DL2000,DL1000)、PCR premix购 自Takara公司,DNA凝胶回收试剂盒、高纯度质粒抽提试剂盒购自Axygen公司,胰蛋白胨、酵母提取物购自OXOID公司(英国),琼脂粉购自 Genehk公司,G418购自 Genview公司,嘌呤霉素购自 Solabio公司,流式破膜固定试剂盒、PE Mouse Anti-Mouse Notch1购自BD公司,盐酸强力霉素购自上海楷洋生物有限公司。
1.2 pcDNA3.1-Notch1真核表达载体的构建
根据 Notch1(NM-017617.3)的功能编码区序列和pcDNA3.1(+)载体的多克隆位点,选择BamHI和XbaI酶切位点作为酶切位点设计引物,引物由上海生物工程有限公司合成Notch1-F:5'CGCGGATCCATGCGCAAGCGCCGGCGGCAGCAT3';Notch1-R:5'ACGTCTAGACACGTCTGCCTGGCTCGG3',下划线部分分别表示BamHI和XbaI的酶切位点。参照总RNA提取试剂盒(Trizol)说明抽提人胎盘组织的总RNA,用RevertAid第一链cDNA合成试剂盒反转录cDNA第一链,PCR扩增Notch1胞内段(NICD)cDNA。PCR反应体系总体积 50μl,其中含模板 cDNA 2μl,2×Premix 25μl,Notch1-F 1μl,Notch1-R 1μl,灭菌双蒸水21μl。PCR反应条件:94℃,预变性 5 min;进入循环:94℃1min,60℃ 90 s,72℃ 90 s,共 30个循环;72℃延伸10min。扩增反应结束后PCR终产物用1%的琼脂糖凝胶电泳检测,并用无水乙醇纯化PCR产物。将带有酶切位点PCR产物用BamH I和XbaI进行双酶切,回收目的基因片段,将 pcDNA3.1(+)载体进行相应的酶切,回收线性化载体。将载体与目的基因按摩尔比1∶5连接过夜,将上述连接产物转化感受态菌 JM109,涂板,,挑选阳性克隆进行 BamH I和XbaI双酶切鉴定并送上海生物公司测序。鉴定正确的克隆命名为pcDNA3.1-Notch1。
1.3 pcDNA3.1-Notch1-EGFP载体的构建
以pEGFP-C1为模板进行PCR扩增EGFP基因,扩增产物为744 bp。EGFP扩增引物为:EGFP-F:5'GGGCCCTCTAGAATGGTGAGCAAGGGCGAGGAG3';EGFP-R:5'GGGCCCGCTAGCGAATTCTTACAGCTCGTCCATGCCGAGAGT 3',下划线部分分别为 XbaI、NheI和EcoRI的酶切位点,EcoRI为引入的验证酶切位点。PCR反应体系40μl,其中含模板质粒 pEGFP-C1 2 μl,Premix 20μl,EGFP-F 1μl,EGFP-R 1μl,灭菌双蒸水16μl。PCR反应条件:94℃,预变性5min;进入三温循环:94℃ 30 s,55℃ 45 s,72℃ 30 s,共 30个循环;72℃延伸10min。将纯化后的PCR产物经XbaI和NheI双酶切产生两个同尾粘末端,回收目的片段。以XbaI酶切 pcDNA3.1-Notch1,DNA凝胶电泳回收载体片段。T4 DNA连接酶连接上述EGFP和线性化pcDNA3.1-Notch1。连接产物转化感受态大肠杆菌JM109细胞,涂板,过夜培养。挑单克隆做菌落PCR,挑选EGFP扩增阳性克隆进一步提取质粒,用EcoR I和BamH I双酶切,DNA凝胶电泳,根据酶切后片段长度判段EGFP的连接方向。鉴定正向连接的克隆命名为pcDNA3.1-Notch1-EGFP。
1.4 pLVX-Notch1-EGFP载体的构建
用EcoR I和 BamH I双酶切 pcDNA3.1-Notch1-EGFP,DNA凝胶电泳并胶回收Notch1-GFP片段。用EcoR I和BamH I双酶切pLVX-Tight-Puro载体,DNA凝胶电泳并胶回收线性化载体。将线性化的pLVXTight-Puro载体和Notch1-EGFP片段用T4连接酶连接,连接产物转化感受态JM109后涂板培养。挑单克隆做菌落PCR,挑选EGFP扩增阳性克隆送生物公司测序。测序正确的载体命名为pLVX-Notch1-EGFP。
1.5 慢病毒包装
包装前24 h,分别接种4~5×106个293T细胞到2个100mm培养皿中。按照慢病毒包装试剂盒(Lenti-XTMHTX Packaging System,Clontech)说明书,分别包装 Tet-On调控系统的反应质粒(pLVX-Tight-Puro-NICD-EGFP)和调控质粒(pLVX-Tet-On Advanced),准备两个 1.5 ml的 EP管 A、B,A管加入557μl的 Xfect Reation Buffer,36μl Lenti-X HTX Packaging Mix,7μg的目的质粒,B管加入592.5μl Xfect Reaction和 7.5μl Xfect polymer,涡旋混匀,将 B管的试剂逐滴加入A管中,涡旋混匀,室温孵育10 min,将A管液体逐滴加到293T细胞中,轻轻晃动培养皿使之混匀,放入培养箱中37℃、5%CO2孵育8 h后换新鲜培养基,再培养36 h收获病毒上清,500×g离心10 min去除细胞碎片。病毒上清可马上用于感染,或分装置于-80℃保存。
1.6 慢病毒感染PC12及抗性筛选
感染前24 h接种2×105cells/well的 PC12细胞于六孔板,病毒感染时细胞50%~70%融合。按照1∶1的比例每孔各加pLVX-Tight-Puro-NICD-EGFP和pLVX-Tet-On Advanced病毒上清400μl,并各加入终浓度为4μg/ml的 polybrene。感染 8~12 h后,吸出原培养基,换为终浓度为 600μg/ml G418和 1.5μg/ml嘌呤霉素培养基进行筛选。之后每3天更换一次抗性培养基,连续筛选14 d,命名该细胞为PC12-Notch1细胞。
1.7 Dox调控Notch1-EGFP的表达
接种1×104cells/well的 PC12-Notch1细胞于24孔板,分为 Dox(+)组和 Dox(-)组,每组设 5个平行样,在Dox(+)组培养基中加入终浓度为500μg/ml Dox,Dox(-)组不加 Dox。观察荧光前进行 DAPI核染色,在培养基中加入 DAPI(终浓度为 50 g/ml),37℃、5%CO2孵育2 h后弃去培养基,PBS洗两遍。分别在 Dox加入后的 0 h、12 h、24 h、36 h和 48 h于荧光倒置显微镜下观察EGFP表达和DAPI核染色,计算EGFP阳性细胞百分比(EGFP阳性细胞数/DAPI阳性细胞数×100%)。
1.8 流式细胞术检测Notch1表达
1.8.1 检测Dox诱导下不同时间点的Notch1表达接种1×104cells/well的 PC12-Notch1细胞于 24孔板,共5组,每组三个平行样,均加入终浓度为500 ng/ml Dox,分别在加入 Dox后的 0 h、12 h、24 h、36 h和48 h收集细胞,按照流式破膜固定试剂盒的步骤处理,上机检测Notch1的表达量及表达Notch1的阳性细胞百分比。独立实验重复三次。
1.8.2 检测不同Dox浓度诱导下的Notch1表达接种1×104cells/well的 PC12-Notch1细胞于 24孔板,分别用不同浓度Dox诱导,终浓度分别为0 ng/ml、100 ng/ml、500 ng/ml、1μg/ml、5μg/ml和 10μg/ml。每个浓度设三个平行样。诱导36 h后收集细胞,按照流式破膜固定试剂盒的步骤处理 PC12-Notch1细胞,上机检测 Notch1的表达量及表达Notch1的阳性细胞百分比。独立实验重复三次。
1.9 统计学分析
所有数据以均数±标准差(±s)表示,统计处理用SPSS 13.0软件进行独立样本t检验。
2 结果
2.1 pcDNA3.1-Notch1载体的鉴定
对重组质粒 pcDNA3.1-Notch1进行 BamHI和XbaI双酶切并进行DNA凝胶电泳,结果显示重组质粒经双酶切后,产生约5.4 kb的 pcDNA3.1(+)载体片段和约2.1kb的Notch1片段(图1)。送测序结果与 Notch1(GenBank:NM-017617.3)序列一致。
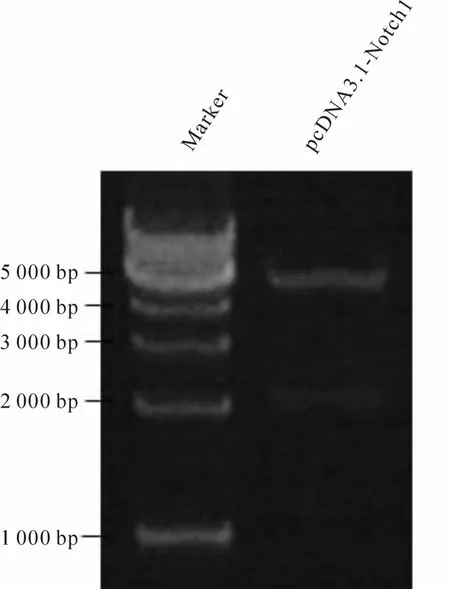
Fig.1 pcDNA3.1-Notch1 was digested with BamHIand XbaI
2.2 pcDNA3.1-Notch1-EGFP载体的鉴定
用EcoR I和 BamH I双酶切 pcDNA3.1-Notch1-EGFP,酶切产物进行1%的琼脂糖凝胶电泳(图2)。酶切产物为 2.8 kb(Notch1+EGFP)和 5.4 kb(pcDNA3.1)两个片段的克隆为正向连接克隆;酶切产物为 2.1 kb(Notch1)和 6.1 kb(pcDNA3.1+EGFP)两个片段的克隆为反向连接克隆。
2.3 pLVX-Notch1-EGFP载体的鉴定
将pLVX-Notch1-EGFP质粒进行EcoR I和BamH I双酶切,酶切产物进行1%的琼脂糖凝胶电泳(图3)。结果证实Notch1-GFP与pLVX-tight-puro载体连接成功。酶切鉴定正确的克隆送公司测序,测序结果与 Notch1(GeneBank:NM-017617.3)序列一致。

Fig.2 pcDNA3.1-Notch1-EGFP was digested with EcoR I and BamH I
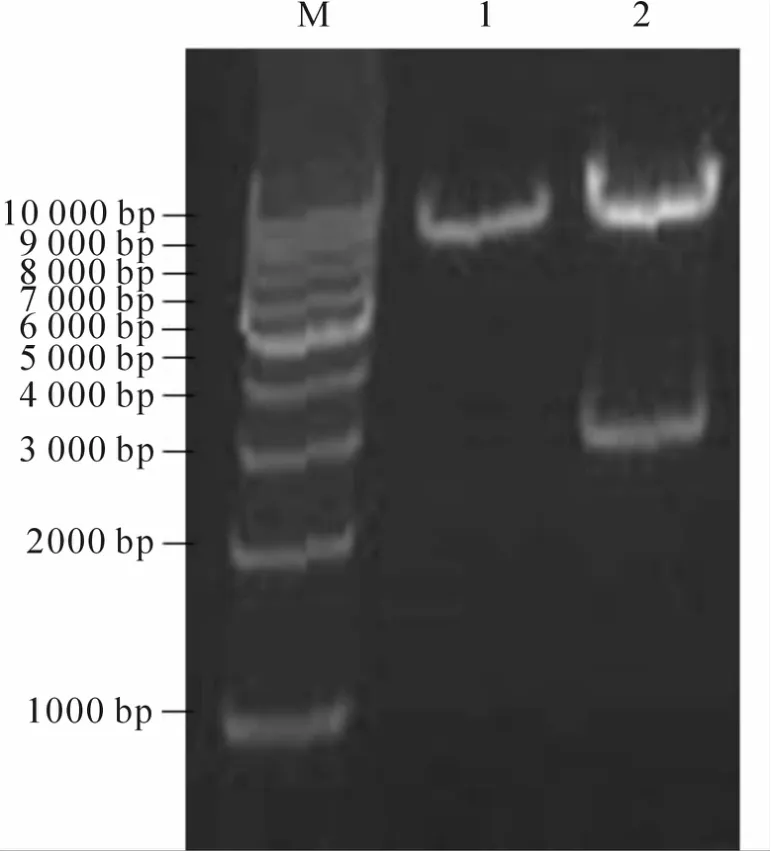
Fig.3 pLVX-Notch1-EGFPwas digested with EcoR Iand BamH I M:DNA Marker;1:Products of pLVX-tight-puro vector by EcoR Iand BamH I digestion;2:Products of pLVX-Notch1-EGFP by EcoR Iand BamH I digestion,which produces 7.2 kb and 2.8 kb fragments
2.4 稳定表达pLVX-Notch1-EGFP的 PC12-Notch1细胞株建立
抗性筛选2周后,在细胞培养基中加入终浓度为 500 ng/ml的 Dox诱导 36 h,Dox(-)对照组无 EGFP表达,Dox(+)组中有EGFP表达。DAPI核染色后计算EGFP阳性细胞百分比,EGFP阳性细胞数大于为90.2%。从图 Merged(DOX+)可以看出,绿色荧光蛋白表达主要集中在细胞核(图4)。
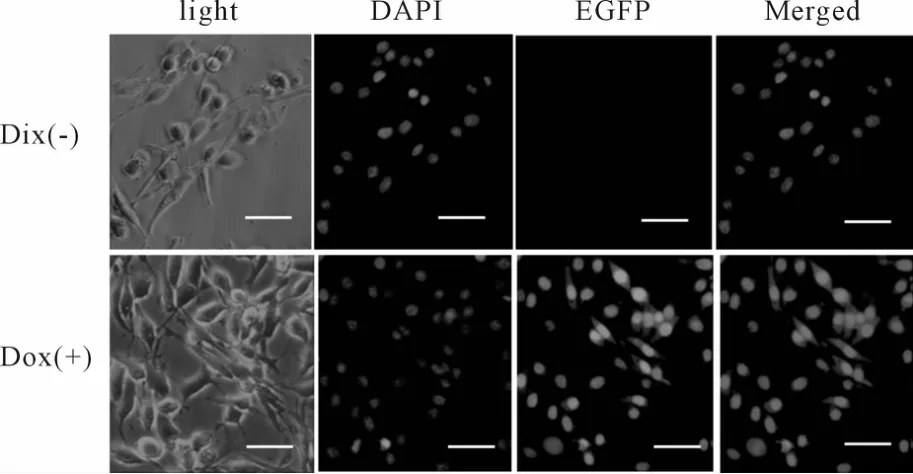
Fig.4 PC12-Notch1 cells under inverted fluorescencemicroscope(×200)
2.5 流式细胞术检测Dox不同诱导时间和不同浓度诱导下Notch1表达
流式细胞术检测 Notch1阳性细胞百分比。Notch1在PC12-Notch1细胞和PC12细胞空白对照组中均维持低表达,Notch1阳性细胞百分比分别为6.1%和 7.2%(P>0.05)。PC12-Notch1细胞经过500 ng/ml的 Dox诱导不同时间(0、12 h、24 h、36 h、48 h)后,Notch1阳性细胞百分比分别为 6.1%、59.7%、66.2%、81.5%、65.5%(图 5A),36 h达到外源性蛋白表达高峰。PC12-Notch1细胞经过不同浓度 Dox(100 ng/ml、500 ng/ml、1μg/ml、5μg/ml、10 μg/ml)诱导 36 h后,Notch1阳性细胞百分比分别为77.4%、81.5%、86.9%、94.8%和 98%(图 5B)。Dox浓度在 10μg/ml及 10μg/ml以下时,细胞均能良好增殖并维持正常细胞形态,显示Dox无明显毒性作用。
3 讨论
Notch信号系统最初是在果蝇的神经系统发育研究中发现的。迄今为止在脊椎动物中共发现4个Notch的同源体,包括 Notchl、Notch2、Notch3和 Notch4(int3)。Notch1受体属于I型膜蛋白,它在不同物种之间(从果蝇到人)以及同一物种的不同成员之间都有高度的结构同源性。人Notchl由胞外亚基(Notch extracellular subunit,NEC)和跨膜亚基(Notch transmembrane subunit,NTM)组成,两个亚基之间通过Ca2+依赖的非共价键结合在一起形成异源二聚体。Notch信号通路的激活需要经过三步酶切过程,在高尔基复合体内被furin样蛋白酶在其S1位点酶切为两条多肽链,即NEC和NTM。配体与Notch1受体结合后促使NEC与NTM发生解离,同时激活后续的S2和S3位点的酶切事件,从而释放了Notch1受体的激活形式(Notch intracellular domain,NICD),转位入细胞核发生核内应答。在本实验中,我们发现当Dox诱导Notch1(NICD)-EGFP融合蛋白在PC12细胞中表达时,绿色荧光主要集中在细胞核,表明该融合蛋白也可转位入细胞核,从而在核内发挥其调控作用。
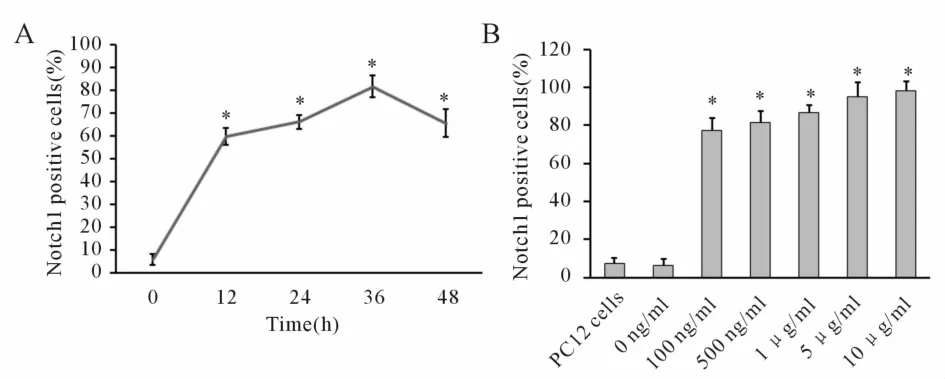
Fig.5 Time and dose dependency between Dox induction and Notch1 expression detected by flow cytometry in PC12-Notch1 cells
本实验采用慢病毒介导的外源性基因表达系统,相对于脂质体等转染方法而言,该系统对于多数细胞均有较高的转染效率。在PC12细胞中,慢病毒感染后再经过抗性筛选,成功获得稳定表达外源性基因Notch1的PC12-Notch1细胞系。该细胞系可以稳定传代并冻存、复苏,为研究Notch1基因在神经细胞的功能提供了良好模型。
PC12-Notch1细胞中Notch1表达受到Dox调控。该调控方式采用Tet-on基因调控系统,该系统由大肠杆菌Tn10转座子中四环素阻遏操纵子的两个部件四环素阻遏蛋白(TetR)和四环素操纵子序列(TetO)与四环素(Tet)及其衍生物(Dox)组成。在没有四环素的情况下,rtTA不能识别TetO,所以外源性基因表达水平为零;而在加入四环素衍生物Dox后,rtTA构像改变,可以与TetO特异的结合,启动下游目的基因表达,并且呈剂量依赖性。本研究在慢病毒介导的Tet-on系统的应答质粒中插入携带Notch1-EGFP融合蛋白,慢病毒包装后感染PC12细胞,建立了PC12-Notch1细胞系。结果发现在PC12-Notch1细胞系中Notch1阳性细胞百分比维持在较低水平,与没有病毒感染的PC12细胞中的Notch水平无明显差异,显示在没有Dox的培养环境里,外源性基因的转入不会引起PC12细胞Notch表达水平的改变。按照 Clontech公司推荐 Dox诱导浓度(500 ng/ml)诱导 PC12-Notch1细胞,发现 Dox作用 12 h后就能检测到Notch1表达显著增高,随着诱导时间延长,Notch1表达逐步增高并在36 h达到顶峰,之后Notch1阳性细胞略有下调,但仍然显著高于Dox调控前。我们进一步研究了不同Dox浓度对Notch1表达的诱导效率,采用的检测时间点是高峰点,即诱导后36 h,结果显示随着Dox浓度增加,Notch1阳性细胞百分比显著增加。在低浓度100 ng/ml Dox作用下,Notch1阳性细胞百分比达到77.4%;之后从100 ng/ml到 10μg/ml Dox浓度,Notch1阳性细胞百分比随着Dox浓度增加而上升的速度变缓。虽然本实验中EGFP和Notch1是融合蛋白并同时在细胞内表达,但在500 ng/ml Dox作用36 h时,EGFP阳性细胞百分比为90.2%(荧光显微镜下计数),显著高于Notch1阳性细胞百分比(流式细胞术检测,81.5%)。由于上述差异可能是流式细胞术检测的敏感性低于绿色荧光计数方法造成,我们认为500 ng/ml Dox可以较好启动Notch1-EGFP融合蛋白的表达,同时该浓度Dox仍在安全无毒范围内,适用于体外诱导实验。
综上所述,本研究成功构建了携带人源性Notch1-EGFP融合蛋白的可调控的慢病毒真核表达载体,建立四环素调控的PC12-Notch1细胞系,并阐明体外调控Notch1在PC12-Notch1细胞中表达的最佳Dox浓度和调控时间。
【参考文献】
[1] Dang T P.Notch,apoptosis and cancer[J].AdvExpMed Biol,2012,727:199-209.
[2] Imayoshi I,Sakamoto M,Yamaguchi M,etal.Essential rolesofNotch signaling inmaintenance ofneural stem cells in developing and adult brains[J].Neurosci,2010,30(9):3489-3498.
[3] Jing L,Jia Y,Lu J,etal.MicroRNA-9 promotes differentiation ofmouse bonemesenchymal stem cells into neurons by Notch signaling[J].Neuroreport,2011,22(5):206-211.
[4] 王建鹏,姚维成,栗世方.下调 Notch-1基因表达抑制胶质母细胞瘤的增殖活性研究[J].中华神经外科杂志,2011,27(2):140-143.
[5] 张家墅,甄海宁,章 翔.Notch信号通路与脑肿瘤研究现状[J].中华神经外科疾病研究杂志,2008,7(6):567-569.
[6] Vilaboa,N.Voellmy R.Regulatable gene expression systems for gene therapy[J].CurrGeneTher,2006,6(4):421-38.
[7] Guruharsha K G,Kankel MW,Artavanis-Tsakonas S.The Notch signalling system:recent insights into the complexity of a conserved pathway[J].NatRevGenet,2012,13(9):654-666.
[8] Lai E C.Notch signaling:control of cell communication and cell fate[J].Development,2004,131(5):965-973.
[9] Li Y,Lau W M,So K F,etal.Caveolin-1 promote astroglial differentiation of neural stem/progenitor cells through modulating Notch1/NICD and Hes1 expressions[J].Biochem BiophysResCommun,2011,407(3):517-524.
[10] Sun Y,Chen X,Xiao D.tetracycline-inducible expression systems:new strategies and practices in the transgenicmouse modeling[J].ActaBiochimBiophysSin(Shanghai),2007,39(4):235-246.
