脆壁克鲁维酵母基因置换技术的建立
2013-02-20曹宇婷卢松冲
李 杰,曹宇婷,徐 欣,张 旭,李 璐,吕 洋,卢松冲
(东北农业大学生命科学学院,哈尔滨 150030)
20世纪80年代后半期,应用DNA同源重组原理,基因置换技术逐渐发展并应用到多种领域。利用基因置换技术,克服随机整合的盲目性和偶然性,能够对细胞染色体进行精确修饰和改造构建目标载体,而目经修饰和改造的基因能进行稳定遗传[1]。目前,基因置换技术已广泛应用于细菌、酿酒酵母、毕赤酵母、丝状真菌和动物等的基因敲除、转基因和基因修饰研究中。克鲁维酵母是天然新鲜牛乳中含有的微生物,来源于克鲁维酵母菌的乳糖酶是一种中性乳糖酶,不仅活性高,其最适pH与天然牛乳的pH(6.6~6.8)最为接近[2],适合工业化处理牛乳和乳清。此外,来源于克鲁维酵母菌的乳糖酶另一优点是在低温时仍具有较好活力,可在低温下分解乳糖,避免乳品加工过程中的腐败菌污染引起牛奶酸败变质。但是,来源于克鲁维酵母的乳糖酶产量并不理想,采用基因工程手段提高乳糖酶表达量是增加乳糖酶产量的有效方法。
本研究以中性乳糖酶生产菌脆壁克鲁维酵母(Kluyveromyces fragilis)为材料,通过构建以脆壁克鲁维酵母乳糖酶基因上游调控区为同源臂的基因置换载体,优化脆壁克鲁维酵母电击转化的条件,试图建立一套有效的脆壁克鲁维酵母基因置换技术,以期为利用精确的基因修饰技术构建高表达中性乳糖酶生产菌奠定基础。
1 材料与方法
1.1 菌种
1.1.1 菌种和质粒
菌株脆壁克鲁维酵母(哈尔滨美华生物技术有限公司提供),质粒pPIC9K(东北农业大学双宝讲师惠赠),质粒pTPlac、pPkan(东北农业大学生命科学学院遗传学研究室构建所得)。
1.1.2 试剂
Pyrobest Taq酶、限制性内切酶、T4DNA连接酶(购自大连宝生物工程公司);DNA胶回收试剂盒(购自北京百泰克生物技术有限公司);其他试剂均为国产分析纯(购自大连宝生物工程公司)。
1.1.3 培养基
YPD:用于酵母培养,主要成分有10 g·L-1酵母提取物,20 g·L-1蛋白炼,20 g·L-1葡萄糖。
YPDG:用于转化子筛选,主要成分有10 g·L-1酵母提取物,20 g·L-1蛋白胨,20 g·L-1葡萄糖,200 mg·L-1G418。
1.2 方法
1.2.1 质粒pPkan的构建和线性化
采用CTAB法提取脆壁克鲁维酵母基因组,以其为模板,利用Plac扩增引物(Sence-Plac:5'TC GCCGATTTGTAACACTCCT 3',Antisence-Plac:5'CTCGTCAATGACCCAGAAGCC 3',3.2 kb)扩增乳糖酶基因调控区Plac,将PCR产物连接到pMD18-T载体上,酶切正确的阳性转化子送交测序。根据pPIC9K的序列(Invirtrogen公司)和PLac序列,对质粒pPIC9K和pTPLac进行Mfe I和Eco R V双酶切,电泳回收载体片段和目的片段,加入T4DNA连接酶连接构建质粒pPkan。电击转化时,用Sal I和Eco R I线性化质粒pPkan。
1.2.2 脆壁克鲁维酵母电击转化条件的优化
1.2.2.1 脆壁克鲁维酵母的活化培养及感受态的制备
挑取脆壁克鲁维酵母的单菌落接种于10 mL YPD液体培养基中,28℃,200 r·min-1,振荡培养过夜。再以1%的接种量转接于100 mL YPD培养基中,28℃培养至OD600=0.5~1.5,4℃。5 000 r·min-1离心5 min,弃去上清,用100 mL冰预冷的无菌水将菌体重悬。4℃,5 000 r·min-1离心10 min,弃去上清,用50 mL冰预冷的无菌水将菌体重悬。4 ℃,5 000 r·min-1离心10 min,再用20 mL,1 mol·L-1山梨醇洗涤 1次,溶于200 μL 1 mol·L-1冰预冷的山梨醇中,以备转化(现做现用)。
1.2.2.2 脆壁克鲁维酵母生长时期对电击转化率的影响
每隔1 h对二次活化的脆壁克鲁维酵母培养液取样测OD600值,绘制生长动力曲线,并取处于OD600为0.5、0.8、1.0、1.5的脆壁克鲁维酵母制备感受态,以1.0 kV电压进行电击转化,电击后培养60 min涂板。
1.2.2.3 电压对脆壁克鲁维酵母电击转化率的影响
固定电容 C=25 μF,电阻=200 Ω,OD600为0.8,以电压分别为0.5、1.0、1.5、2.0、2.5 kV进行电击转化,电击后培养60 min涂板。
1.2.2.4 电击后涂布前培养时间对转化的影响
OD600为0.8,电压1.5 kV,电击后分别培养0、30、60、90、120 min进行涂板。
1.2.3 限制内切酶介导法在酵母电击转化中的应用
OD600为0.8,电压1.5 kV,取200 μL感受态细胞加入质粒pPkan混合均匀。分别加入0、5、8、10限制性内切酶Eco R I和Sau I,冰浴30 min后,以电压1.5 kV进行电击转化,转化后培养60 min,吸取200 μL涂布。

图1 重组质粒pPKan的构建Fig.1 Construction of plasmid pPKan
1.2.3 转化子鉴定
根据pPIC9K的序列和pTPLac序列分析,应用软件Premier 5.0进行引物设计,引物对Genekan用以检测kan基因的插入(Genekan-Sense:5'GATGG TCGGAAGAGGC 3',Genekan-Antisense:5'CCAGT AGTAGGTTGAGGC 3',600 bp),引物对 Upkan、Downkan分别用以检测上下游同源臂的同源插入(Upkan-Sense:5'TTTGCCCACCCTCTTG 3',Upkan-Antisense:5'TTGCCCGCTAATGCTA 3',1.5 kb;Downkan-Sense:5'CAAGTATGTCTGCCTGTATT 3',Downkan-Antisense:5'AGCCTCCAAGTTCGTAT 3',1.0 kb)。
2 结果与分析
2.1 脆壁克鲁维酵母电击转化条件的优化
2.1.1 脆壁克鲁维酵母生长时期对电击转化率影响
每隔一小时对二次活化的脆壁克鲁维酵母培养液取样测OD600值,得到生长动力曲线(见图2)。
脆壁克鲁维酵母的不同生长期对电击转化率表现出较大影响,当OD600为0.8时转化率最高,为1.92×102转化子·μg-1DNA,继续培养反而使转化率下降(见图3),说明处于对数生长期中前期的细胞可以获得较高的转化效果。

图2 脆壁克鲁维酵母生长动力曲线Fig.2 Curve of growth of Kluyveromyces fragilis
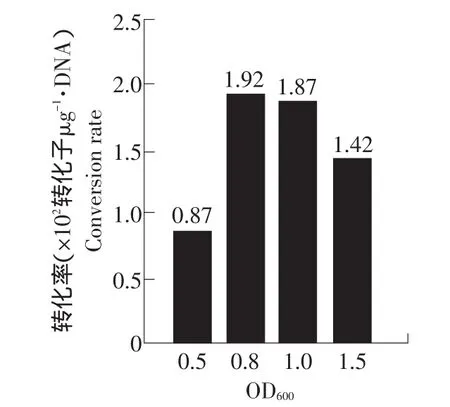
图3 OD600 对脆壁克鲁维酵母电击转化的影响Fig.3 Ecffect of OD600 on electroporation Kluyveromyces fragilis
2.1.2 电压对脆壁克鲁维酵母电击转化率的影响
电场强度是电击转化中最敏感的参数,直接影响转化率[3],而电容C=25 μF,电阻=200 Ω比较适合微生物电击转化[4],试验中采用0.2 cm的小电击槽,固定其他参数,调节电压从0.5~2.5 kV,得到电场强度为2.5~12.5 kV·cm-1。当电场强度为7.5 kV·cm-1时转化率最高,达到2.37×102转化子·μg-1DNA,继续增加电压,转化率反而下降(见图4)。这种转化频率随电压的变化说明了较低的电压不足以引起细胞变化为易于接受外源DNA的生理状态,而过高的电压则会对细胞造成较大的伤害,细胞膜产生不可逆破裂[3],使细胞死亡率过高,有效转化率降低。

图4 电压对脆壁克鲁维酵母电击转化的影响Fig.4 Eeffect of voltage on electroporation of Kluyveromyces fragilis
2.1.3 电击后涂布前培养时间对转化的影响
在相同的试验条件下,电击后加入培养基立即涂布和培养不同时间后涂布。电击后培养120 min时转化率最高。这种转化频率随转化后培养时间的变化说明了由于标记基因转入后,需要较长时间表达其功能。电击后培养120 min后涂板相较于培养90 min后涂板,转化率并没有显著的提高(见图4),造成这种结果的原因可能由于培养90 min已经足够时间使电击后的脆壁克鲁维酵母进行恢复,延长时间仅促进酵母的分裂繁殖,考虑到缩短操作时间和培养过程中的细胞分裂不利于转化子筛选等因素[5],以培养90 min后涂板为宜。
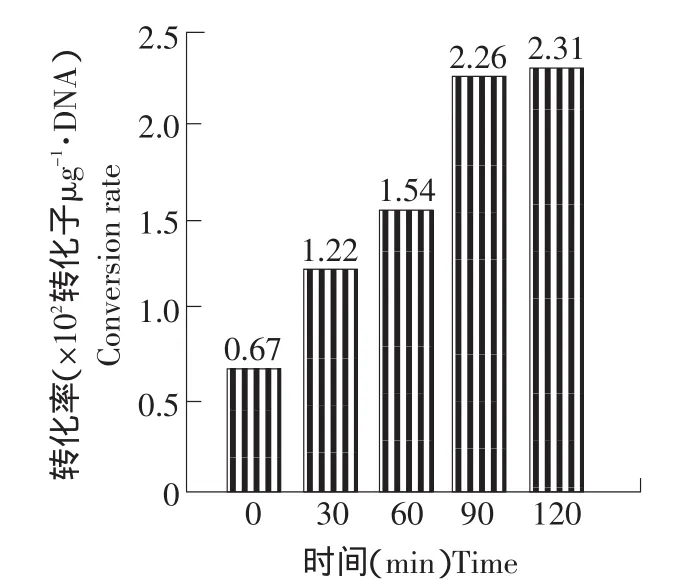
图5 电击后涂布前培养时间对转化的影响Fig.5 Eeffect of culture time on electroporation of Kluyveromyces fragilis
2.2 限制内切酶介导法在酵母电击转化中的应用
限制酶介导转化是在限制酶存在下用线性化质粒DNA转化真菌,外源基因可以插在基因组的相应酶切位点,从而获得转化子。REMI被广泛应用于动植物病原真菌以及其他真菌的转化中[6-8],普遍可获得较高的转化率[9-10]。该方法首先在酵母菌中由Schiestl建立,转化率比直接转化提高了7倍[11]。而在本试验中,加入限制内切酶后,与未加入限制内切酶的对照相比转化率仅相差0.2×102转化子·μg-1DNA,并未显著提高转化率,可能原因是Schiestl建立的REMI法建立在原生质体转化法的基础上,而本试验中采取的电击转化法已经具有较高的转化率,因此REMI法并未对转化率有显著的提高。
2.3 转化子鉴定
PCR检测结果见图6~8。

图6 PCR扩增检测kan基因Fig.6 Detection of kan gene by PCR

图7 PCR扩增检测上游同源臂的插入优点Fig.7 Detection of insertion site of upstream homologous arm by PCR

图8 PCR扩增检测下游同源臂的正确插入Fig.8 Detection of insertion site of downstream Homologous arm
电击转化法是目前转化效率最高的转化方法之一,但采用电击转化法时,发生假阳性的频率也是最高的[12]。因此,仅仅利用抗性或利用营养缺陷型对重组子进行筛选是不够的,有必要对重组子进行二次筛选,最简便有效的二次筛选法为PCR法。随机选取10个转化子,用引物对kan基因进行PCR扩增验证,可扩增出600 bp条带的即为阳性转化子,试验结果证明80%的转化子为阳性转化子(见图6)。用引物对Upkan、Downkan对阳性转化子的整合位点进行PCR扩增验证,上下游分别可扩增出1.5 kb、1.0 kb条带即为进行了同源重组的转化子,试验结果证明阳性转化子中87.5%进行了同源重组(见图7~8),获得较高的同源重组率。
3 讨论与结论
基因置换技术在研究基因功能和实现基因精确缺失有广泛应用,但成功率低[13],提高同源重组率和转化率是解决问题关键。同源臂长度对同源重组率有重要影响,在酿酒酵母系统中,当两端同源臂为30 bp时,同源重组率可高达80%[14]。而在非常规酵母系统中,如多形汉逊酵母,即使两端同源臂为1 kb时,同源重组率仅为50%[15]。脆壁克鲁维酵母为非常规酵母,试验选择1 kb同源区作为同源臂,以期获得较高的同源重组率。对脆壁克鲁维酵母的电击转化条件进行优化。不同的转化条件对转化率有较大影响,研究可知,制备感受态时脆壁克鲁维酵母的生长时期和电击转化时电场强度对转化率影响较大,合适条件可以提高转化率,以OD600为0.8的脆壁克鲁维酵母为受体、电场强度为7.5 kV·cm-1、电击后培养时间为90 min,可获得较高的转化率,最高转化效率为2.37×102转化子·μg-1DNA。而采用REMI法,在电击转化的同时加入限制性内切酶,对电击转化转化率无显著影响。
试验的下一步将以乳糖酶基因上游调控区缺失的脆壁克鲁维酵母为载体,对其启动子的上游激活序列及信号肽进行基因工程改造。对其启动子的上游激活序列的改造,在提高乳糖酶的生物活性的基础上,并未有改变脆壁克鲁维酵母乳糖酶本身的基因序列,避免以往表达系统表达外源基因时,分泌表达的蛋白不能正确折叠和修饰加工等问题引起乳糖酶的活性降低甚至失活;并尝试已在毕赤酵母中成功表达了外源蛋白的信号肽α-Factor信号肽和菊粉酶基因信号肽INU,以期获得高表达中性乳糖酶的分泌型表达载体。
[1]Capecchi M R.Altering the genome by homologous recombination[J].Science,1989,224:1288-1292.
[2]郭杰炎,毛敏伟,孙玉华,等.酵母乳糖酶对牛乳乳糖酶水解作用的研究[J].食品与发酵工业,1991(3):19-22.
[3]William J Dower,Jeff F Miller,Charles W Ragsdale.High efficiency transformation of E.coli by high voltage electroporation[J].Nucleic Acids Research,1988,16(13):6127-6145.
[4]Valentina Ganeva,Bojidar Galutzova,Justin Teissieb.Influence of glucose and other substrates on electric field and polyethylene glycol-mediated transformation of intact yeast cells[J].FEMS Microbiology Letters,1994,121(2):159-164.
[5]刘媛美.酵母细胞的电击高频转化[J].暨南大学学报,1996,16(3):6-9.
[6]Yun S H,Turgeon B G,Yoder O C.REMI-induced mutants of Mycosphaerella zeae-maydis lacking thepolyketide PM-toxin are deficient in pathogenesis to corn[J].Physiol Mol Plant Pathol,1998,52:53-66.
[7]Redman R S,Rodriguez R J.Factors affecting the efficient transformation of Colletotrichum species[J].Exp Mycol,1994,18:230-246.
[8]Garnand K,Nelson M A.The erect of DNA structure and restriction enzymes on transformation efficiencies in Neurospora crassa[J].Fungal Genet Newslett,1995,42:29-31.
[9]Aden K,William F L.Tagging developmental genes in Dictyostelium by restriction enzyme-mediated integration of plasmid DNA[J].Genetics,1992,89(18):8803-8807.
[10]Kim S,Sonq J,CHoi H T.Genetic transformation and mutant isolation in Ganoderma lucidum by restriction enzyme-mediated integration[J].FEMS Microbiol Lett,2004,233(2):201-204.
[11]Schiestl R H,Petes T D.Integration of DNA fragments by illegitimate recombination in Saccharomyces cerevisiae[J].PNAS,1991,88(17):7585-7589.
[12]Choi J,Kim E E,YI Park.Expression of the active human and duck hepatitis B virus polymerases in heterologous system of Pichia methanolica[J].Antivir Res,2002,55:279-290.
[13]刘建忠,敖敬,田为宇,等.基因敲除技术研究进展[J].化学与生物工程,2006,23(2):4-6.
[14]Hua S B,Qiu M,Chan E,et al.Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast[J].Plasmid,1997,38(2):91-96.
[15]Celedonio G,Genman P,Paula T,et al.One-step,PCR-mediated,gene disruption in the yeast Hansenula polymorpha[J].Yeast,1999,15:1323-1329.
