3-O-乙酰基-4,6-二-O-苄基-1,2-环丙烷葡萄糖的合成*
2012-11-21徐立炎邵华武
徐立炎, 邵华武
(1. 中国科学院 成都生物研究所 天然产物研究中心,四川 成都 610041;2. 中国科学院 研究生院,北京 100049)
1,2-环丙烷糖广泛用于2-C-支链吡喃糖和氧杂七环糖苷的制备[1,2]。Zou W等[3,4]发现甘露糖的1-位丙酮基在中等碱性条件下可以进攻2-位磺酸酯离去基团,生成乙酰基取代的1,2-环丙烷葡萄糖。这类环丙烷糖可作为糖基供体,与单糖、氨基酸和各类醇在Lewis酸催化下发生糖苷化反应选择性地得到α-或β-糖苷[5,6]。
本文以葡萄糖为原料,经5步反应制得苄基烯糖(2);2经Ferrier重排制得2,3-不饱和碳苷(3);3经高锰酸钾羟基化反应制得3-C-(4,6-二-O-苄基-α-D-吡喃甘露糖基)丙酮(4);4经选择性磺酰化制得3-C-(4,6-二-O-苄基-2-O-甲基磺酰基-α-D-吡喃甘露糖基)丙酮(5);5经乙酰基羟基保护制得3-C-(3-O-乙酰基-4,6-二-O-苄基-2-O-甲基磺酰基-α-D-吡喃甘露糖基)丙酮(6);6在碱性条件下关环合成了3-O-乙酰基-4,6-二-O-苄基-1,2-环丙烷葡萄糖(7, Sceheme 1),其中新化合物3~7的结构经1H NMR,13C NMR和HR-MS表征。
1 实验部分
1.1 仪器与试剂
Brucker-600 MHz型核磁共振仪(CDCl3为溶
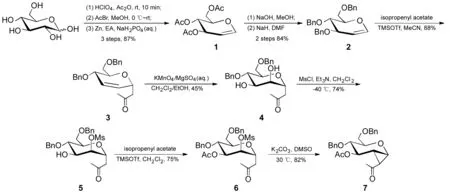
Scheme 1
剂,TMS为内标);BioTOF Q型质谱仪。
所用试剂均为市售分析纯。
1.2 合成
(1) 乙酰化葡萄烯糖(1)的合成[7]
在圆底烧瓶中加入含结晶水的D-葡萄糖20.0 g(111 mmol),醋酐100 mL,于0 ℃搅拌均匀后加1滴HClO4,缓慢升至室温,反应30 min;冰水浴冷却下加入乙酰溴33 mL(446 mmol),缓慢滴加甲醇18 mL(444 mmol),滴毕,于室温反应4 h。加入乙酸乙酯(EA)200 mL,冰水200 mL,分液,水层用乙酸乙酯(200 mL)萃取,合并有机层,加入饱和NaH2PO4水溶液400 mL,锌粉80 g,于室温剧烈搅拌7 h。抽滤,滤液分液,水层用乙酸乙酯(100 mL)萃取,合并有机层,加入饱和NaHCO3溶液200 mL,于室温剧烈搅拌15 min;分液,有机层用无水Na2SO4干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=3 ∶1]纯化得无色糖浆1 26.2 g,产率87%。
(2)2的合成
在反应瓶中加入1 1.00 g(3.67 mmol)的甲醇(5 mL)溶液和NaOH 150 mg(3.75 mmol),搅拌于室温反应30 min;减压浓缩,残余物用DMF(10 mL)溶解,加入NaH 530 mg(22 mmol),反应5 min;于0 ℃加入溴化苄2.2 mL(18.5 mmol),缓慢升至室温,反应2 h。加入二氯甲烷35 mL,依次用水(3×30 mL),饱和食盐水(30 mL)洗涤,无水MgSO4干燥,浓缩后经硅胶柱层析(洗脱剂:A=10 ∶1)纯化得白色固体21.28 g,产率84%。
(3)3的合成
在反应瓶中加入2 1.12 g(2.69 mmol)的乙腈(27 mL)溶液和醋酸异丙烯酯 420 μL(4.82 mmol),搅拌下于0 ℃滴加TMSOTf(三甲基硅烷基三氟甲磺酸酯)18 μL(0.1 mmol),滴毕,反应5 min。加适量三乙胺淬灭反应,浓缩,残余物经硅胶柱层析(洗脱剂:A=4 ∶1)纯化得浅黄色糖浆30.87 g,产率88%;1H NMRδ: 7.39~7.26(m, 10H), 5.97~5.92(m, 1H), 5.89~5.81(m, 1H), 4.72~4.66(m, 1H), 4.63~4.57(m, 2H), 4.53(d,J=12.1 Hz, 1H), 4.49(d,J=11.6 Hz, 1H), 4.00~3.93(m, 1H), 3.85~3.77(m, 1H), 3.70~3.65(m, 2H), 2.89(dd,J=15.8 Hz, 8.0 Hz, 1H), 2.57(dd,J=15.8 Hz, 5.8 Hz, 1H), 2.19(s, 3H);13C NMRδ: 206.6, 138.2, 138.1, 130.7, 128.4, 127.9, 127.8, 127.6, 126.0, 73.4, 71.8, 71.1, 69.8, 69.3, 69.0, 47.0, 30.8; ESI-HR-MSm/z: Calcd for C23H26O4Na{[M+Na]+} 389.172 3, found 389.173 3。
(4)4的合成
在反应瓶中加入3 816 mg(2.23 mmol)的二氯甲烷(4 mL)溶液和乙醇40 mL,搅拌下于0 ℃滴加高锰酸钾700 mg(4.43 mmol)和MgSO4500 mg的水(7 mL)溶液,滴毕,缓慢升至室温,反应4 h。用NaHSO4饱和水溶液淬灭反应,用二氯甲烷(3×50 mL)萃取,合并有机层,用无水MgSO4干燥,浓缩,残余物经硅胶柱层析(梯度洗脱剂:A=1 ∶1~0 ∶1)纯化得浅黄色浆4400 mg,产率45%;1H NMRδ: 7.39~7.25(m, 10H), 4.65(d,J=11.7 Hz, 1H), 4.61(d,J=11.8 Hz, 1H), 4.57(d,J=11.8 Hz, 1H), 4.54(d,J=11.9 Hz, 1H), 4.34(dd,J=13.0 Hz, 6.6 Hz, 1H), 3.94~3.90(m, 1H), 3.86~3.82(m, 1H), 3.76~3.71(m, 2H), 3.70~3.66(m, 2H), 2.81~2.68(m, 2H), 2.19(s, 3H);13C NMRδ: 207.1, 137.9, 137.4, 128.5, 128.0, 127.9, 127.8, 76.9, 74.0, 73.7, 73.0, 70.4, 70.3, 69.9, 69.6, 45.4, 30.7; ESI-HR-MSm/z: Calcd for C23H28O6Na{[M+Na]+} 423.177 8, found 423.178 4。
(5)5的合成
在反应瓶中加入4350 mg(874 μmol)的二氯甲烷(9 mL)溶液和三乙胺 730 μL(5.27 mmol),搅拌下于-40 ℃滴加甲基磺酰氯(MsCl) 135 μL(1.74 mmol),滴毕,反应30 min。加二氯甲烷30 mL,依次用水(3×30 mL),饱和食盐水(30 mL)洗涤,无水MgSO4干燥,浓缩,残余物经硅胶柱层析(洗脱剂:A=1 ∶1)纯化得浅黄色糖浆5310 mg,产率74%;1H NMRδ: 7.37~7.22(m, 10H), 4.81(dd,J=4.7 Hz, 3.3 Hz, 1H), 4.67(d,J=11.5 Hz, 1H), 4.65~4.60(m, 2H), 4.58(d,J=11.5 Hz, 1H), 4.54(d,J=11.9 Hz, 1H), 4.10(dd,J=7.2 Hz, 3.2 Hz, 1H), 3.79~3.68(m, 4H), 3.09(s, 3H), 2.80(dd,J=16.2 Hz, 7.9 Hz, 1H), 2.70(dd,J=16.2 Hz, 5.8 Hz, 1H), 2.20(s, 3H);13C NMRδ: 205.3, 137.8, 128.6, 128.4, 128.0, 127.8, 80.0, 76.3, 74.3, 73.8, 73.7, 69.7, 69.3, 69.1, 44.0, 38.7, 30.5; ESI-HR-MSm/z: Calcd for C24H30O8SNa{[M+Na]+} 501.155 4, found 501.155 4。
(6)6的合成
在反应瓶中加入5 165 mg(345 μmol)的二氯甲烷(3.5 mL)溶液和醋酸异丙烯酯46 μL(418 μmol),搅拌下于0 ℃滴加TMSOTf 10 μL(55 μmol),反应5 min。用三乙胺淬灭反应,浓缩后经硅胶柱层析(洗脱剂:A=2 ∶1)纯化得浅黄色糖浆6135 mg,产率75%;1H NMRδ: 7.38~7.25(m, 8H), 7.21~7.16(m, 2H), 5.16(dd,J=8.6, 3.1 Hz, 1H), 4.89(t,J=3.2 Hz, 1H), 4.64(d,J=11.4 Hz, 1H), 4.62~4.56(m, 2H), 4.55~4.50(m, 2H), 3.90(t,J=8.1 Hz, 1H), 3.76~3.66(m, 3H), 3.08(s, 3H), 2.88(dd,J=17.2 Hz, 6.6 Hz, 1H), 2.83(dd,J=17.1 Hz, 7.2 Hz, 1H), 2.20(s, 3H), 2.04(s, 3H);13C NMRδ: 204.7, 170.3, 137.9, 137.7, 128.5, 128.4, 127.9, 127.8,77.4, 74.5, 74.4, 73.6, 73.0, 70.7, 68.6, 43.2, 38.6, 30.6, 20.9; ESI-HR-MSm/z: Calcd for C26H32O9SNa{[M+Na]+} 543.165 9, found 543.166 4。
(7)7的合成
在反应瓶中加入670 mg(134 μmol)的二甲亚砜(1.4 mL)溶液和碳酸钾100 mg(724 μmol),搅拌下于30 ℃反应6 h。加二氯甲烷20 mL,依次用水(3×20 mL)与饱和食盐水(20 mL)洗涤,无水MgSO4干燥,浓缩,残余物经硅胶柱层析(洗脱剂:A=4 ∶1)纯化得浅黄色糖浆747 mg,产率82%;1H NMRδ: 7.38~7.24(m, 10H), 5.16~5.08(m, 1H), 4.66(d,J=11.7 Hz, 1H), 4.58(d,J=11.8 Hz, 1H), 4.57~4.51(m, 2H), 3.84(dd,J=7.0 Hz, 1.7 Hz, 1H), 3.80(dd,J=10.5 Hz, 5.4 Hz, 1H), 3.63~3.57(m, 2H), 3.63~3.57(m, 1H), 2.56~2.52(m, 1H), 2.28(s, 3H), 2.02(s, 3H), 1.83(t,J=5.7 Hz, 1H);13C NMRδ: 204.7, 169.9, 137.8, 137.7, 128.4, 127.9, 127.8, 127.7, 74.0, 73.9, 73.5, 72.9, 69.2, 68.7, 59.9, 32.5, 31.2, 26.1, 21.0; ESI-HR-MSm/z: Calcd for C25H28O6Na{[M+Na]+} 447.177 8, found 447.177 4。
2 结果与讨论
在1,2-环丙烷葡萄糖合成过程中,高锰酸钾羟基化反应有许多文献[8,9]报道,且产率较高。本文将该方法应用于合成3时,产率偏低。对于选择性磺酰化反应,随着温度的升高副产物增多。孔繁祚等[10]曾报道选择性磺酰化方法,本文将该方法应用于合成4时,反应复杂。
7的稳定性比3,4,6-三-O-苄基-1,2-环丙烷葡萄糖差,可能是由3-乙酰氧基的吸电子效应增大引起的。7的3-位乙酰化,便于进一步去保护修饰,而6-位苄醚也可以转换成其他官能团,这样就可以实现3,4,6-位羟基分别被不同的基团保护,在寡糖及其他具有生物活性糖类衍生物的合成中更具实用价值,同时为糖类化合物选择性保护和官能团转化提供了新的方法。
[1] Cousins G S, Hoberg J O. Synthesis and chemistry of cyclopropanated carbohydrates[J].Chemical Society Reviews,2000,29(3):165-174.
[2] Yu M, Pagenkopf B L. Recent advances in donor-acceptor(DA) cyclopropanes[J].Tetrahedron,2005,61(2):321-347.
[3] Shao H, Ekthawatchai S, Wu S H,etal. 2-C-branched glycosides from 2′-carbonylalkyl 2-O-Ms(Ts)-C-glycosides.A tandem SN2-SN2reaction via 1,2-cyclopropanated Sugars[J].Organic Letters,2004,6(20):3497-3499.
[4] Shao H, Ekthawatchai S, Chen C S,etal. 1,2-Migration of 2′-oxoalkyl group and concomitant synthesis of 2-C-branchedO-,S-glycosides and glycosyl azides via 1,2-cyclopropanated sugars[J].The Journal of Organic Chemistry,2005,70(12):4726-4734.
[5] Tian Q, Xu L, Ma X,etal. Stereoselective synthesis of 2-C-acetonyl-2-deoxy-D-galactosides using 1,2-cyclopropaneacetylated sugar as novel glycosyl donor[J].Organic Letters,2009,12(3):540-543.
[6] Tian Q, Dong L, Ma X,etal. Stereoselective synthesis of 2-C-branched(acetylmethyl) oligosaccharides and glycoconjugates:Lewis acid-catalyzed glycosylation from 1,2-cyclopropaneacetylated sugars[J].Journal of Organic Chemistry,2011,76(4):1045-1053.
[7] 邵华武,赵晋忠. 一种葡萄烯糖的制备方法[P].CN ZL 200 710 048 471.x,2008.
[8] Tsang R, Fraser-Reid B. A route to optically active trichothecane skeleton by bisannulation of a pyranose derivative[J].The Journal of Organic Chemistry,1985,50(23):4659-4661.
[9] Hunter L, Slawin AMZ, Kirsch P,etal. Synthesis and conformation of multi-vicinal fluoroalkane diastereoisomers[J].Angewandte Chemie International Edition,2007,46(41):7887-7890.
[10] Kong F, Du J, Wu H. Selective esterification of methyl 4,6-di-O-benzyl-α-D- mannopyranoside[J].Carbohydrate Research,1986,147(2):337-341.
