通脉抗栓颗粒质量标准研究
2012-11-09李成网
张 洁 白 娟 李成网*
(安徽省医学科学研究院,安徽 合肥 230061)
通脉抗栓颗粒是根据临床经验方开发的六类中药新药,已获国家药品食品监督管理局颁发的临床批件(批件号:2007L05140),由当归、川芎、葛根、人参叶等中药组成,具有益气养阴,活血通脉等功效。用于脑血栓症见头晕目眩,头痛,肢体麻木或强急,气短乏力,便干便秘,口干口渴,舌质红或质暗淡等气阴两虚, 瘀阻脑络证。采用薄层色谱法对其中的当归、川芎、麦冬、人参叶进行定性鉴别,采用HPLC法对葛根中的有效成分葛根素进行含量测定,可有效控制通脉抗栓颗粒的质量。
1 仪器、试药与样品
Waters 600型高效液相色谱仪(美国Waters公司);UV 996型紫外检测器;AD-2型百万分之一电子天平(美国PE公司);BP210S型万分之一电子天平(德国赛多利斯);AS3120型超声清洗器(天津奥特赛恩斯);葛根素为对照品(批号0752-9806供含量测定用);人参皂苷Rb1对照品(批号704—8903);人参皂苷Re对照品(批号754-9002);人参皂苷Rg1对照品(批号0703-200119);当归对照药材(批号120927-200310);川芎对照药材(批号120918-200306);麦冬对照药材(批号121013-200607)均购于中国药品生物制品检定所。甲醇、磷酸为色谱纯,水为重蒸水,其他试剂均为化学纯。通脉抗栓颗粒由安徽省华康医药科技开发有限责任公司提供(规格:每袋装8g)。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 当归川芎的鉴别[1]
取本品15g,研细,加乙醚150ml浸渍1h后,超声处理5min,滤过,滤液挥干,残渣加醋酸乙酯0.5mL使溶解,作为供试品溶液。另取当归对照药材和川芎对照药材各1g,分别加乙醚50mL,浸泡1h,超声处理5min,滤过,滤液挥干,残渣加醋酸乙酯2mL使溶解,分别作为对照药材溶液。照薄层色谱法试验,分别吸取当归和川芎对照药材溶液3μL,供试品溶液10μL,分别点于同一硅胶G薄层板上,以正已烷-醋酸乙酯(9∶1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显两个相同颜色的荧光斑点。阴性对照无干扰。见图1。
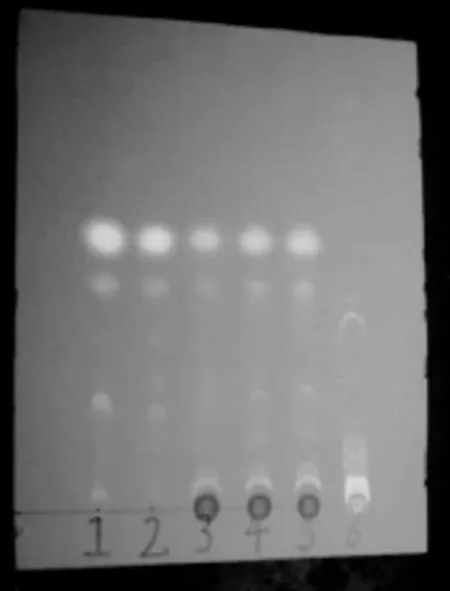
图1 当归川芎鉴别薄层色谱图
2.1.2 麦冬的鉴别[2]
取鉴别(1)项下乙醚提取后的药渣,加乙醇100mL回流1h,滤过,滤液回收乙醇至干,加3%硫酸溶液10mL置沸水浴加热回流2h,用3%氢氧化钠溶液调pH至中性,水浴蒸干,残渣加氯仿2mL使溶解,作为供试品溶液,另取麦冬对照药材2g,加乙醇20mL回流1h,同法制成对照药材溶液。照薄层色谱法试验,吸取对照药材溶液10μL和供试品溶液10μL,分别点于同一硅胶G薄层板上,以正己烷-醋酸乙酯(1∶1)为展开剂,展开,取出,晾干,喷以10%硫酸溶液,于105°C加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。阴性对照无干扰。见图2。
2.1.3 人参叶的鉴别[3,4]
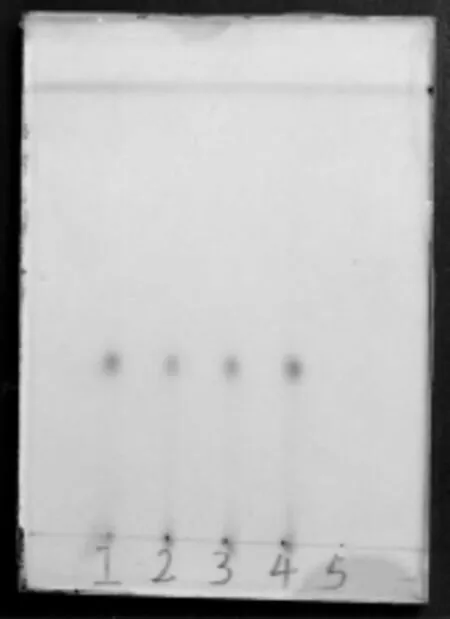
图2 麦冬鉴别薄层色谱图
取本品5g,研细,加甲醇50mL,超声处理20min,滤过,滤液蒸干,残渣加水25mL使溶解,滤过,滤液加水饱和正丁醇提取3次,每次25mL,合并正丁醇提取液,用正丁醇饱和的氨试液洗3次,每次25mL,弃去氨试液,正丁醇液水浴蒸干,残渣加甲醇2mL使溶解,作为供试品溶液。另取人参皂苷Rb1、Re、Rg1对照品,分别加甲醇制成每1mL各含0.5mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取供试品溶液5~10μL,对照品溶液3~5μL,分别点于同一硅胶G薄层板上,以正丁醇-醋酸乙酯-水(4∶1∶5)的上层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至色斑清晰,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。阴性对照无干扰。见图3。

图3 人参叶鉴别薄层色谱图
2.2 葛根素的含量测定
葛根为方中君药,其主要成分为葛根素。用高效液相色谱法测定中药制剂中葛根素的含量的报道很多[5,6]。在参考文献的基础上,为了有效地控制本品的质量,建立了本品的葛根素的高效液相色谱法的测定法,经方法学考察,本法简便、准确、可靠、重现性好,可有效控制本品质量,其方法学考察如下:
2.2.1 色谱条件与系统适用性试验
2.2.1.1 对照品溶液的制备
精密称取葛根素对照品适量,加30%乙醇制成每1mL含60μg的溶液,即得。
2.2.1.2 供试品溶液的制备
取本品,研细(过4号筛),取0.5g,精密称定,置具塞锥形瓶中,精密加入30%乙醇50mL,密塞,称定重量,超声处理30min,放冷,再称定重量,用30%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.2.1.3 阴性对照溶液的制备
取缺葛根的阴性制剂,按供试品溶液制备项下的方法依法制备阴性对照溶液。
2.2.1.4 色谱条件的选择
色谱柱:Shim-Pack CLC ODS柱(150×4.6mm,5μm);流动相:甲醇-0.1%磷酸溶液(24∶76);检测波长:250nm;柱温:30℃;流速:1.0mL/min。在此条件下,葛根素与其它成分均能很好地分离,供试品中葛根素保留时间与对照品一致,且阴性对照无干扰,结果见图4。

图4 对照品、样品、阴性样品HPLC色谱图
2.2.5 线性关系考察
精密吸取葛根素对照品溶液(0.21744mg/mL)1.0、2.0、3.0、4.0、5.0mL分别置10mL量瓶中,加30%乙醇稀释至刻度,摇匀。分别精密吸取20μL,依次注入液相色谱仪中,按上述色谱条件测定,峰面积分别为:1516068、2931786、4409670、5889355、7336747。以葛根素的浓度为横坐标,相应的峰面积积分值为纵坐标,计算,得线性回归方程为:A=67140C+37047 r=0.99997(n=5)。表明葛根素在21.744~108.72μg/mL范围内呈良好的线性关系。
2.2.6 供试品精密度试验
取按供试品溶液制备法制备的供试液(批号040805),按上述色谱条件注入液相色谱仪,连续进样5次,记录葛根素峰面积分别为:5179674、5275425、 5276877、5392464、5317029,RSD=1.46%,表明精密度良好。
2.2.7 稳定性试验
将按供试品溶液制备法制备的供试液(批号040805)于0、1、2、4、6、8h按上述色谱条件重复测定一次,记录葛根素峰面积分别为:5463520、5417945、5489535、5464131、5471071、5561982,RSD=0.87%。表明供试液在8h内稳定。
2.2.8 重复性试验
取同一批号样品(批号040805)共5份,按供试品溶液制备法制备供试液,精密量取该溶液和对照品溶液各20mL,按上述色谱条件注入液相色谱仪,记录色谱图,计算葛根素含量分别为65.5728mg/袋,65.7864mg/袋,64.1368mg/袋,65.0384mg/袋,64.9488mg/袋,RSD=0.98%。表明方法重复性良好。
2.2.9 回收率试验
精密量取已知含量(65.0968mg/袋)的样品适量,分别精密加入葛根素对照品适量,按正文含量测定项下的方法依法制备供试品溶液并测定,结果见表1。

表1 回收率试验结果(n=6)
2.2.10 样品测定
对3批中试样品按供试品溶液制备方法制备样品溶液,分别精密吸取样品溶液和对照品溶液各20mL,注入液相色谱仪,按外标法以峰面积计算含量,结果见表2。

表2 样品含量测定结果
3 讨 论
3.1 本方为中药复方制剂,采用薄层色谱鉴别方法对方中的每一味中药均进行了鉴别,并进行了阴性对照试验,结果其方法简便且专属性强,可有效控制本品的质量。
3.2 关于葛根中葛根素的含量测定方法,文献报道很多,但在本制剂中未见报道,对流动相进行了选择,先后比较了甲醇-水(25∶75)、甲醇-水(22∶78)、甲醇-0.1%磷酸溶液(22∶78)、甲醇-0.1%磷酸溶液(24∶76)。结果以甲醇-0.1%磷酸溶液(24∶76)为流动相最佳。
3.3 对供试品溶液制备的超声提取时间也做了考察,分别比较了超声处理10、20、30、40min的效果。结果表明,超声提取30min已能提取完全,因此超声提取时间选择30min为佳。
[1]中华人民共和国卫生部药典委员会编.中华人民共和国药典[S].2000年版一部.北京:化学工业出版社,2000:433.
[2]余伯阳.不同麦冬假叶树皂甙元的含量比较[J].中国药科大学学报,1987,18(2):117.
[3]中华人民共和国卫生部药典委员会编.中华人民共和国药典[S].2000年版一部.北京:化学工业出版社,2000:6.
[4]苗明三,李振国.现代实用中药质量控制技术[M].北京:人民卫生出版社,2000:36.
[5]杨小平.RP-HPLC法测定感冒清热颗粒中葛根素的含量[J].药物分析杂志 2001,21(5):331.
[6]刘建峰.HPLC法测定舒心片中葛根素含量[J].西北药学杂志,2000,15(5):1990.
