叠加对比气相色谱法在中药含量测定中的应用
2012-09-18徐国防
徐国防 李 爽 纪 佳
(郑州人民医院,河南 郑州 450003)
中药包括中草药和中成药,在我国的应用已经有上千年的历史,在世界上也受到了越来越多的关注和开发应用。过去,中药的定量只要称量一下药材的重量即可,现在越来越需要测定出其中有效成分的精确含量,但中药成分特别复杂多样,在测定一些挥发性成分时,采用气相色谱法常难以寻找到合适的内标。本文在叠加对比法理论[1]的基础上,结合气相色谱法的特点,设计了叠加对比气相色谱测定中药成分的试验方案。

表1 线性回归数据和最低检测限
叠加对比法[1],即在色谱图中选取与待测成分色谱峰的保留时间和峰面积相近的稳定色谱峰作为参比峰(相当于内标峰),用待测成分峰面积与参比峰之比代替峰面积的绝对值的定量方法。计算公式如下:
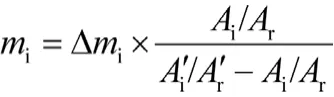
式中:Ai为待测成分的峰面积;Ai′为增加待测成分纯物质△mi后,再进样,待测成分的峰面积;Ar为参比峰的峰面积;Ar′为增加待测成分纯物质△mi后,再进样,测得的参比峰的峰面积。
1 仪器与试药
Agilent 6890N型气相色谱仪配FID检测仪(美国安捷伦公司);ChemStation工作站(美国安捷伦公司);BS110 S电子天平(德国Sartorius公司);泌尿宁颗粒(自制,批号:20080901,20080923,20081009);五味子甲素(批号:0764-200006)、五味子乙素(批号:0706-200003)均购自中国药品生物制品检定所;试剂均为分析纯,水为重蒸水。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱为:HP-5 Phenyl Methyl Siloxane capillary column (30.0 m×0.32mm,0.50μm);载气:高纯度N2;载气流速:1.2mL/min;检测器:氢焰离子化检测器(FID);燃气为:氢气(30mL/min)和空气(300 mL/min);检测器温度:280℃,进样口温度:230℃;进样量:1μL;分流比:1∶50;升温程序为:150 ℃维持3 min后,以10℃/min的速度升至210℃,接着以5 ℃/min的速度从210℃升至240℃,再以10 ℃/min的速度升至280℃,维持5min。在上述色谱条件下测定,各待测成分色谱峰、参比峰之间及与其他共存成分色谱峰之间能够达到基线分离,各待测成分色谱峰、参比峰的理论塔板数均不低于3000。色谱图见图1A。
2.2 溶液制备
2.2.1 供试品溶液的制备
取泌尿宁颗粒12.0g,照《中国药典》2010年版一部 附录X D挥发油测定法提取,用正己烷萃取,再用无水硫酸钠脱水处理后,用正己烷定容至25mL,得储备供试液。将1.0mL储备供试液用正己烷稀释至10mL,用微孔滤膜滤过,即得供试品溶液。
2.2.2 对照品溶液的制备
精密称取五味子甲素、五味子乙素对照品适量,混合,用正己烷溶解,得到浓度分别为0.0600和0.240 mg/mL的混合储备标准液。另取上述储备供试液1.0mL,再加入1.0mL上述混合储备标准液,用正己烷稀释并定容至10mL,用微孔滤膜滤过,即得对照品溶液。
2.3 线性试验
取已知含量的颗粒剂12g,精密称定,置圆底烧瓶中,照“供试品溶液的制备”方法制成储备供试液。精密量取5份该储备供试液各0.5mL分别置于10mL的量瓶中,在其中4份溶液中依次加入0.5、1.5、2.5、3.5mL储备标准液。将以上5份溶液加正己烷稀释至刻度,混匀,进样分析,每份两针,记录色谱图,求每份样品中待测成分峰面积与参比峰峰面积比的平均值,再将此峰面积比的平均值与溶液中待测成分的浓度进行线性回归,结果见表1。逐步稀释溶液,测定最低检测限,结果见表1。

图1 供试品溶液(图A)和对照品溶液(图B)色谱图峰:1-五味子甲素;2-五味子乙素;3-参比峰
2.4 精密度和回收率试验
取供试品溶液,重复进样6次,记录色谱图,计算五味子甲素、乙素色谱峰峰面积与参比峰峰面积比的相对标准偏差。结果见表2。由表2可见方法的精密度良好。

表2 精密度测定试验结果
取已知含量的泌尿宁颗粒样品共9份,每份6.0 g,分别精密称定,加入8.0,10.0,12.0mL混合储备标准液各3份。将所得的9份样品分别置于圆底烧瓶中,照上述方法制备供试品溶液和对照品溶液,测定加对照品后的含量。计算回收率(recovery):

式中,mtotal是测得的总量,moriginal 是未加对照品时已知样品中的含量,mspiked是加入的对照品的量。结果五味子甲素、五味子乙素的回收率分别为:96.7% (RSD 3.6%),99.3%(RSD 4.8%),表明方法的准确度基本良好。
2.5 样品测定
取3批不同批号的样品,按“供试品溶液的制备”和“对照品溶液的制备”项下操作制备供试品溶液和对照品溶液。在上述色谱条件下测定,分别精密吸取供试品溶液和对照品溶液各1μL,进样测定,记录色谱图。分析供试品溶液色谱图和对照品溶液色图谱,选择与待测成分色谱峰保留时间和峰面积相近的稳定色谱峰作为参比峰,按叠加对比法计算颗粒剂中五味子甲素等两种成分的含量:

式中:mi为待测成分i的含量,单位mg•g-1;Ci为混合储备标准液中i成分的浓度,单位mg•mL-1;Ai为供试品溶液待测成分(i)的峰面积,Ai′为对照品溶液i成分峰面积;Ar为供试品溶液参比峰的峰面积;Ar′为对照品溶液参比峰的峰面积;W为样品称样量,单位g。测定结果见表3。

表3 样品含量测定结果
3 讨 论
叠加法的理论研究和应用较早[2,3],但限于技术等原因,应用范围不广。随着仪器分析技术的发展和复杂样品分析工作的需要,叠加对比法开始应用于分析领域[4,5],孙国祥教授等还对毛细管电泳叠加对比法的测定误差进行了理论推导[6]。叠加对比法用相对峰面积替代了峰面积的绝对值,因此可以克服外标法中进样量不准确、仪器波动等所带来的定量误差。该法还巧妙的利用了样品中原有成分的色谱峰作为参比峰,避免了内标法在分析复杂样品时较难找到合适的内标物或者色谱峰过于密集,无处可插入内标峰的困惑。总之,叠加对比法虽然不加内标物,却具有内标法的优点,尤其适用于分析成分复杂的样品,如中药等。
根据参比峰的选择原则,一般可以在进样多次后选取与待测成分色谱峰的保留时间和峰面积相近且稳定的色谱峰作为参比峰。参比峰只需要在同一批测定供试品溶液和对照品溶液中保持相对的稳定(峰面积和保留时间的变化仅是由于进样量不准确、仪器波动等偶然因素引起的,不包括参比成分不稳定等引起的变化)即可,而不必要在所有的样品中均保持稳定。经过一定的试验设计,甚至还可以选取不同的待测成分(即已知成分)色谱峰相互作为参比峰。
[1] 孙毓庆,王延琮.现代色谱法及其在药物分析中的应用[M].北京:科学出版社,2005:279-280.
[2] 寇登民,云希勤,李艳红,等.气相色谱叠加定量方法的研究[J].分析化学,1992,20(5):597-599.
[3] 王静华,王宏伟.叠加定量气相色谱法测定吹苯残渣中的三苯含量[J].化学工程师,1997,3(60):54-55.
[4] 孙国祥,苗菊茹,王宇,等.毛细管电泳叠加对比法测定阿片粉中吗啡的含量[J].色谱,2002,20(1):69-70.
[5] 孙国祥,孙毓庆,李阳,等.毛细管电泳叠加对比法测定复方降压片中5组分含量[J].沈阳药科大学学报,2002,19(4):265-268.
[6] 孙国祥,邓湘昱,万月生,等.毛细管电泳叠加对比法的误差分析[J].中南药学,2004,2(5):266-268.
