误诊为多发性肌炎的脂质沉积性肌病——附3例报告☆
2012-09-17管玉青谢作善郑卉潘速跃
管玉青谢作善 郑卉 潘速跃
脂质沉积性肌病(lipid storage myopathy,LSM)是脂质代谢过程中的酶或肉碱等缺乏,影响了肌肉纤维内的脂质代谢,导致异常增多的脂滴在肌纤维内堆积而引起的肌病,表现为近端肌无力、肌痛、运动不耐受等,临床上极易被误诊。我院近2年来经肌肉活检确诊LSM患者3例,现报告如下:
1 资料与方法
1.1 资料例1,女,21岁,学生,因反复四肢及颈部疼痛、无力5年余,加重5 d于2009年9月入院。患者于2004年3月无诱因出现行走时双大腿疼痛,渐出现双下肢无力,蹲位站立及上楼梯困难,头下垂。5月中旬出现双臂上抬无力,症状进行性加重,6月初出现吞咽困难,四肢肌肉压痛明显。查体:语音低沉,吞咽困难;垂头;四肢肌张力低,肌力:双上肢近端0级,远端3级,双下肢近端0级,远端1级,四肢肌肉压痛明显,四肢腱反射消失,病理征阴性。化验示:肌酸激酶(creatine kinase,CK)5592 U/L;肌电图示肌源性损害。诊断为多发性肌炎(polymyositis,PM),予甲基强的松龙冲击治疗,病情略好转,数日后又加重,呼吸肌无力,予呼吸机维持呼吸。复查CK 11070 U/L,继续治疗2个月后好转出院。激素减量停用,肌力渐好转,但不及正常人。4年间反复出现四肢、颈部疼痛、乏力,口服激素可缓解,不能参加大学军训及体育课,上楼梯需休息。2008年9月后肌无力次数增多,激素疗效较差,2009年8月受凉后病情加重,再次入院。查体:咀嚼无力,饮水呛咳,颈软,抬头费力,双上肢近端肌力3级,远端肌力4级,双下肢肌力 2+ 级。化验示 CK 1570 U/L,CK-MB 42.51 U/L,肌红蛋白202 ng/mL。予甲基强的松龙冲击治疗5 d,效果不佳,行左肱二头肌活检。
例2:男,25岁,文员,因四肢疼痛、力弱3个月于2010年7月就诊。查体:四肢肌力5级,双下肢肌肉压痛。肌电图示肌源性损害,CK 4647 U/L。诊断为PM,予强的松30 mg每天口服,1个月后症状减轻,自行停药。2010年9月复诊,仍觉肌肉疼痛,以双腓肠肌疼痛为著。复查CK1908 U/L。行左腓肠肌活检。
例3:女,20岁,无业,因反复四肢疼痛、无力2年余于2010年12月入院。在外院诊断为PM,予激素治疗后好转,但运动耐力差。病情反复,1年内复发2~3次,入院前数月肌无力再次加重,激素治疗效果差,改服中草药治疗,症状渐加重,入院前3 d开始完全卧床。查体:双侧咬肌无力,余颅神经正常,肢体肌力:颈伸肌、颈屈肌3级,三角肌肌力2级,肱二头肌、肱三头肌3级,上肢远端5级;髂腰肌1级,股四头肌3级,股二头肌2级,胫骨前肌、腓肠肌4+级,感觉正常,腱反射消失,病理征阴性。EMG检查示肌源性损害,血CK 4147 U/L。行右肱二头肌活检。
3例患者过去史、家族史均无特殊;例3自幼挑食,不喜食肉、蛋类,例1、2饮食无特殊。
1.2 方法 骨骼肌标本快速冰冻,8 μm切片,行HE、改良 Gomori三色(MGT)和 NADH、SDH、COX、ORO、PAS染色。采集患者空腹血、尿标本送日本MILS实验室北京分部,行血脂酰肉碱及尿有机酸检测。
2 结果
病理及代谢检查结果: 3例患者肌肉病理改变相似,HE染色均表现为肌纤维大小不等,大量肌纤维内见类圆形空泡,部分空泡融合(图1A),未见炎细胞浸润。改良Gomori染色未见不整红边纤维(ragged red fiber,RRF),油红 O 染色见肌纤维内大量红染的脂滴沉积 (图1B)。病理改变符合LSM。例1:丙氨酸、苏氨酸、缬氨酸高值,多种脂酰肉碱比值增高,游离肉碱未见异常,结合尿检查结果,考虑为高乳酸血症或继发性改变;例2:多种氨基酸浓度升高,多种脂酰肉碱浓度增高,游离肉碱未见异常,尿中乳酸排泄明显增高;例3:多种脂酰肉碱浓度增高,尿代谢检查提示戊二酸尿症。
治疗及转归:3例患者确诊后均予低蛋白、低脂肪、高碳水化合物饮食,口服维生素B2、左卡尼汀等药物治疗,症状改善明显,其中例1与例3均在治疗后1周内下床步行,例2的肌痛症状亦在短期内完全缓解。3例患者肌酶谱检查均恢复正常。
3 讨论
肌细胞内的脂肪代谢是一个有多种酶参与的复杂过程,LSM是一个复杂的综合征而非单一病变。肉碱或肉碱棕榈酰转移酶缺乏、多种酰基辅酶A脱氢酶缺乏、线粒体呼吸链功能障碍等都可导致能量代谢障碍和大量脂质在肌细胞内沉积[1-4]。
LSM的临床特点:急性或亚急性起病,病前可有受凉、感染等诱因,典型表现为近端肌无力、运动不耐受,可伴有肌痛,症状可以有波动性;肌电图提示肌源性损害,部分可合并神经源性损害;血清肌酶可正常或明显升高,部分病人伴有呕吐等胃肠道症状。LSM患者常有颈伸肌无力,导致患者呈垂头状,本文中例1与例3均有此表现,有作者提出颈肌、脊旁肌和咀嚼肌早期受累可能是LSM的临床特征[5];LSM患者常有挑食,不喜食肉、蛋、鱼类,本文中例3有此表现。
LSD中骨骼肌内脂肪代谢障碍,造成能量产生不足,导致肌无力、运动不耐受的临床表现,同时,肌细胞内环境的改变、膜的通透性增加,导致CK升高,伴有细胞结构破坏、肌细胞坏死。激素对部分LSM有明显疗效,临床症状和肌酶可在短期内恢复,原因在于激素有促进代谢的作用,还可以稳定细胞膜、减少肌细胞的破坏[6]。激素的治疗反应使得LSM极易被误诊为PM、重症肌无力及慢性吉兰-巴雷综合征等[2-4]。
LSM的诊断金标准是肌肉病理检查,肌细胞中出现大量脂质沉积时可确诊。需注意与PM、线粒体肌病所伴随的继发性脂质沉积相鉴别,认真观察光镜下脂质沉积性改变与炎细胞浸润、不整边红纤维等改变之间的关系是鉴别要点,必要时须结合临床表现及电镜下改变进行分析[6-7]。国内至今报道LSM共300余例,随着规范的骨骼肌活检及染色方法的普及,LSM的报道越来越多。
LSM的发病机制:温冰等的研究提示LSM在中国的发病率可能高于其他国家,该研究还发现国人中的核黄素反应性LSM大多是由ETFDH基因突变导致的轻型多脂酰辅酶A脱氢缺陷(multiple acyl-CoA dehydrogenation deficiency,MADD) 所致[8]。来自台湾的汉族华人LSM的基因研究也得出了相似的结果[9],并提出血中多种脂酰肉碱浓度增高是提示MADD的敏感生化指标。本文中3例患者血代谢检查均发现多种脂酰肉碱浓度或比值增高。近期又有多个大规模的报道证实ETFDH基因突变导致的MADD是导致中国人LSM的主要原因,且突变热点可能有地域差别[10-12]。
对于PM的诊断,迄今为止仍沿用Bohan和Peter在 1975年提出的标准(B/P 标准)[13-14]:①肢带肌和颈肌对称性无力,在数周到数月间逐渐进展,可伴有吞咽困难和呼吸肌受累;②肌肉活检可见肌纤维坏死、吞噬、再生现象,束周肌萎缩,肌纤维大小不等,炎性渗出等;③肌酶升高;④EMG示短时程低波幅的多相运动单位电位,肌纤颤电位、正锐波、插入电位或复杂重复放电。以上四项中有三项符合可诊断PM,若伴有特征性皮疹可诊断皮肌炎(dermatomyositis,DM)。随着对炎性肌病的研究不断深入,这一标准的不足之处也显现出来:区别PM和DM仅靠有无皮疹;不能区分PM与包涵体肌炎(IBM);未列出各型肌炎肌的特异性病理改变;未明确PM与DM或其他导致肌纤维坏死的病变的主要鉴别点;未纳入新的病理学诊断指标。应用B/P标准,若不行病理检查,绝大部分的LSM患者都将被误诊为PM。2004年,欧洲神经肌肉疾病中心和美国肌肉研究协作组提出了新的特发性炎性肌病分类诊断标准[15]。对比新标准与B/P标准中PM的诊断指标可发现:临床表现方面:新标准强调了发病年龄(≥18岁)和肌无力的分布特点(对称性、三角肌无力重于腕/指屈肌,髂腰肌无力重于股四头肌/胫骨前肌;颈屈肌无力重于颈伸肌)。辅助检查方面除电生理诊断指标外还强调了骨骼肌MRI和肌炎特异性抗体的诊断价值;病理方面,提出肌内膜炎细胞浸润、包绕并侵入非坏死肌纤维作为确诊PM的依据,同时强调了可排除PM诊断的肌肉病理改变(如镶边空泡、束周肌萎缩、肌营养不良改变等)。应用新的诊断标准可避免PM的过度诊断,减少代谢性肌病的误诊。
Dalakas在其综述中总结了DM和PM诊断中应注意的问题,笔者认为值得学习和借鉴[16]:①在诊断PM时应排除包涵体肌炎、中毒性肌病、坏死性肌病和肌营养不良,因为根据B/P诊断标准不能对区分PM与这些疾病;②PM是一种较罕见的疾病,包涵体肌炎较其更常见(未包括中国人的流行病学资料);③肌内膜炎症并非PM所特有,也见于肌营养不良、中毒性、代谢性肌病;④做过针极肌电图检查的肌肉在1个月内不应行肌肉活检;⑤对临床表现为疲劳和血清转氨酶或乳酸脱氢酶增高的患者,一定要进行CK检查,以排除肌肉源性的转氨酶增高;⑥PM活动期必有肌无力症状,临床表现为肌痛而肌力正常者不能诊断PM;⑦当肌活检不能显示原发性炎症 (即CD8阳性T细胞侵入组织相容性抗原1表达阳性的非坏死肌纤维)时,PM的诊断不确凿;⑧如果针对PM的治疗仅能降低CK而不能改善肌力,应该对诊断重新评价。
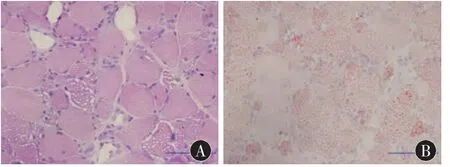
图1 肌活检示肌纤维内大量空泡(图1A,HE染色),ORO染色显示大量脂滴沉积(图 1B),标尺长度:100 μm。
对临床考虑为PM的患者,不要盲目应用激素治疗,应积极进行骨骼肌活检,明确诊断。对病理表现支持LSM的患者,应进行血、尿代谢指标筛查,明确代谢障碍的环节,以指导对因治疗。
[1] 李伟,焉传祝,吴金玲,等.脂质沉积性肌病42例临床治疗和预后随访[J].中华神经科杂志,2007,40(4):229-231.
[2] 骆翔,熊昊,霍江涛,等.脂质沉积性肌病的诊断与鉴别诊断[J].脑与神经疾病杂志,2007,15(2):84-86.
[3] 陈兴泳,胡伟,孟祥武.脂质沉积性肌病临床特征(附10例报告)[J].中国神经精神疾病杂志,2008,34(11):675-676.
[4] 邵素君,沈光莉,韩爽.脂质沉积性肌病2例报告[J].中国神经精神疾病杂志,2004,30(2):100.
[5] 王勤周,焉传祝,吴金玲,等.核黄素反应性脂质沉积性肌病的临床和病理特征[J].临床神经病学杂志,2005,18(5):357-359.
[6] 陈琳,郭玉璞,任海涛,等.貌似多发性肌炎的脂质沉积性肌病病理改变[J].中华神经科杂志,2001,34(2):81-83.
[7] 刘璐,笪宇威,贾建平.线粒体脑肌病合并脂质沉积性肌病1例报告并文献复习[J].北京医学,2007,29(10):590-592.
[8]Wen B,Dai T,Li W,et al.Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations[J].J Neurol Neurosurg Psychiatry,2010,81(2):231-236.
[9]Lan MY,Fu MH,Liu YF,et al.High frequency of ETFDH c.250G > A mutation in Taiwanese patients with late-onset lipid storage myopathy[J].Clin Genet,2010,78(6):565-569.
[10]王韵,赵丹华,洪道俊,等.核黄素反应性脂质沉积性肌病20个家系的电子转移黄素蛋白脱氢酶基因存在热点突变[J].中华神经科杂志,2011,44(5):309-313.
[11]奚剑英,卢家红,赵重波,等.脂质沉积性肌病35例的临床特点及电子转移黄素蛋白脱氢酶基因突变分析[J].中华神经科杂志,2011,44(5):314-320.
[12]Wang ZQ,Chen XJ,Murong SX,et al.Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G > A[J].J Mol Med,2011,89(6):569-576.
[13]Bohan A,Peter JB.Polymyositis and dermatomyositis(first of two parts)[J].N Engl J Med,1975,292(7):344-347.
[14]Bohan A,Peter JB.Polymyositis and dermatomyositis (second of two parts)[J].N Engl J Med,1975,292(8):403-407.
[15]Hoogendijk JE,Amato AA,Lecky BR,et al.119th ENMC international workshop:trial design in adult idiopathic inflammatory myopathies,with the exception of inclusion body myositis,10-12 October 2003; Naarden,The Netherlands.[J].Neuromuscul Disord,2004,14(5):337-345.
[16]Dalakas MC,Hohlfeld R.Polymyositis and dermatomyositis[J].Lancet,2003,362(9388):971-982.
