C'散点图:一种描述氨基酸结构差异的新方法
2011-12-31唐鹤云杨啸林胡俊峰张正国
唐鹤云 杨啸林 胡俊峰 张正国*
(徐州医学院医学影像学院,徐州 221004)2(中国医学科学院基础医学研究所,北京协和医学院基础学院,北京 100005)
引言
蛋白质的空间结构信息都包含在其氨基酸序列中[1]。研究氨基酸几何结构的影响因素,对理解蛋白质独特空间结构的形成具有重要意义[2-3]。同时,氨基酸几何结构的变化规律也是蛋白质结构预测的理论基础[4-5]。目前,研究氨基酸几何结构的方法主要采用二面角(Φ 和 Ψ)[6-8]或 τ[9],Φ 表示氨基氮原子(N)和 α碳原子(Cα)间的空间角。Ψ表示 α碳原子(Cα)和羧基碳原子(C')间的空间角,而τ则是把 Φ和 Ψ结合在一起,计算的是 N、Cα和C'这3个原子间形成的空间角,这些方法通过计算骨架原子形成的两个相邻肽平面的空间角,描述氨基酸的空间结构。这些二面角也可以通过 Ramachandran图进行可视化[10]。但是空间角度比较抽象,并且很难量化地计算相同氨基酸残基在不同结构环境中的结构差异性。
笔者在研究多肽片段中氨基酸结构的实践中,首次提出了C'散点图这种研究氨基酸几何结构的新方法,在应用中体现出了独特的效率和便利,下面将详细介绍这种描述氨基酸结构的新方法。
1 方法
对于多肽片段中任一氨基酸而言,第i个残基的骨架原子 Ni、Cαi和分布在两个相邻的肽平面上(见图 1)。Ni和 Cαi与前一个氨基酸的 Cαi-1、、Oi-1这 3 个原子形成一个肽平面,而 Cαi、氧原子Oi和则与后一个氨基酸的Ni+1和 Ci+1处于同一个肽平面,Cαi是这两个相邻肽平面的轴心。目前,主要用这两个肽平面形成的二面角来描述氨基酸骨架的结构。

图1 转换的参考坐标系Fig.1 The referred coordinate for transformation
在同一个肽平面中,各原子的相对位置是非常固定的。因此,可以用一个肽平面上的原子相对于相邻肽平面的位置,描述这两个平面的空间位置关系。根据骨架原子Ni、Cαi和的关系,选择相对于前一个肽平面的坐标来描述氨基酸的结构。
由于蛋白质结构数据库中各原子的坐标参考系是不同的,所以在运用原子进行氨基酸结构比较前,目标氨基酸中的原子坐标首先要进行转换,使其坐标的原点、方向一致,才具有可比性。选择的前一个肽平面作为参考平面,与原子所在同一个氨基酸的Ni和Cαi以及前一个氨基酸的Cαi-1、Oi-1在这个参考平面上。由于同一个肽平面上的原子分布固定,根据三点确定一个平面的原则,选择前一个氨基酸的 Cαi-1、、Oi-1这 3个原子作为坐标转换的参考。
具体的转换方法可以分为以下3个步骤。
步骤1:以前一个原子的 Cαi-1作为新的坐标原点,也就是把原来的坐标原点平移到 Cαi-1,那么平移后所有原子的坐标变为
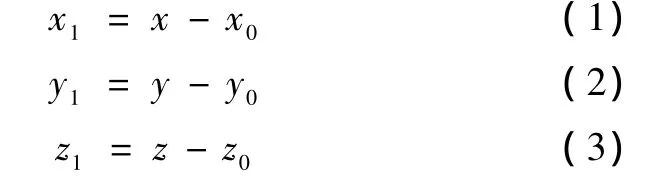
式中,(x,y,z) 为各原子的原始坐标值,(x0,y0,z0)为 Cαi-1的原始坐标,(x1,y1,z1)为各原子的新坐标值。
步骤 2:以 Cαi-1指向 C'i-1作为新的 X 轴。因Cαi-1已经是原点,所以这一步可以分为两个子步骤。首先,整个坐标系绕X轴旋转,直到落在X-Y平面上;然后,再绕Z轴旋转,直到落在X轴上。
步骤2-1:使整个坐标系绕X轴旋转,直到落在X-Y平面上,此时原子与X-Y平面的夹角为
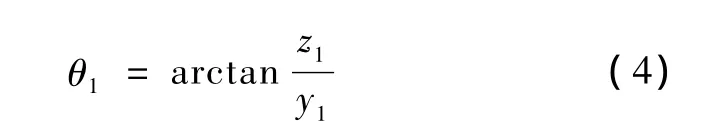
需旋转的角度为
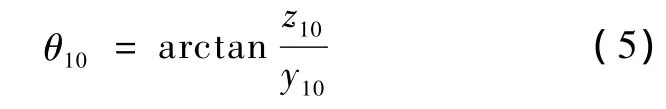
旋转完成后,原子与X-Y平面的夹角为
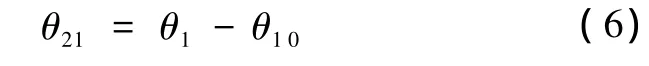
这时各原子的坐标变为
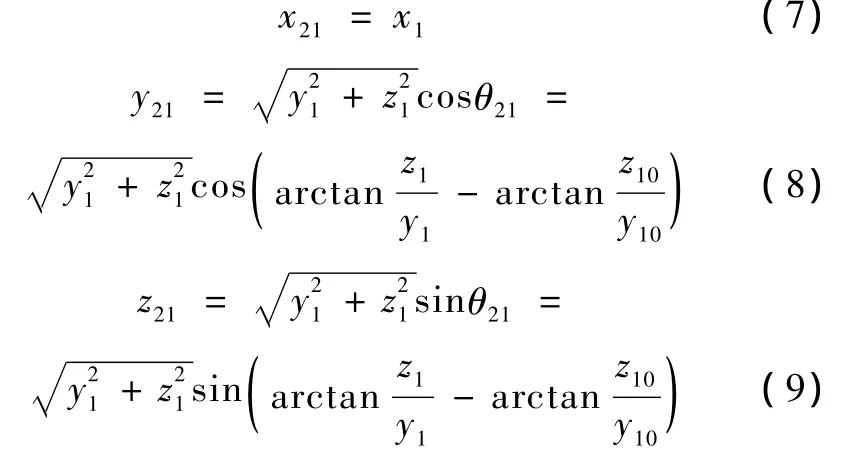
式中,(x1,y1,z1)为各原子在第一步后的坐标值,(x10,y10,z10)为在步骤 1 后的坐标值,(x21,y21,z21)为各原子此时的新坐标值。

旋转的角度为
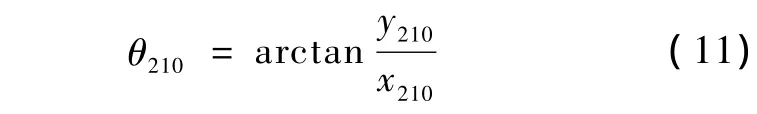
旋转完成后各原子与X-Z平面的夹角变为
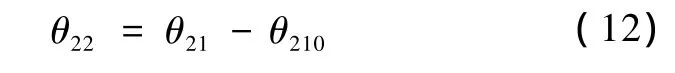
此时各原子的新坐标为
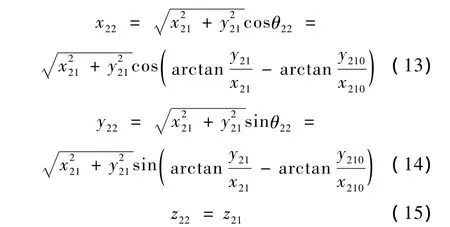
式中,(x21,y21,z21)为各原子在步骤2-1后的坐标值,(x210,y210,z210)为 C'i-1在步骤 2-1 后的坐标值,(x22,y22,z22)为各原子在步骤2-2后的新坐标值。
将步骤2-1和步骤2-2合并,那么步骤2完成后,各原子的坐标为

步骤 3:把 Cαi-1、、Oi-1作为新的X-Y平面。前两步完成后,Cαi-1已经是新的原点,已经在X轴上,因此这一步的任务是绕X轴旋转,直到 Oi-1落在 X -Y平面上。
此时,各原子与X-Y平面的夹角为
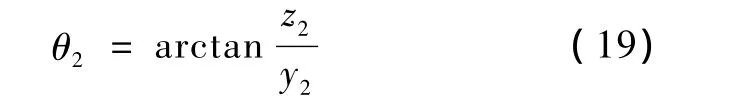
旋转的角度为
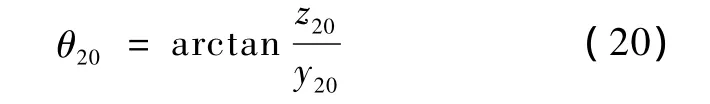
旋转完成后各原子与X-Y平面的夹角变为

那么各原子的坐标将变为

式中,(x2,y2,z2)为各原子在步骤2后的坐标值,(x20,y20,z20)为 Oi-1在步骤 2 后的坐标值,(x3,y3,z3)为各原子此时的新坐标值。
经过上述的坐标转换,新的坐标系是由目标氨基酸的前一个残基的 Cαi-1、和 Oi-1这 3 个原子作为参考而建立的,所以原子的三维坐标就包含了目标氨基酸的骨架结构信息,同时也反映了与前一个氨基酸残基的相对位置关系。相同氨基酸残基的原子在三维坐标图中的分布(散点图)情况,就可以反映这个氨基酸的结构变化情况。
运用C'散点图,可以非常直观地展示各氨基酸残基在多肽片段中结构变化的情况。从蛋白质结构数据库(PDB)可以下载所有目前已知三维结构的蛋白质数据,截取相同组成的多肽片段,就可以运用C'散点图这个方法来分析氨基酸在不同环境下的结构差异。
2 结果
蛋白质结构数据库(Protein Data Bank,http://www.rcsb.org)是一个开放的蛋白质结构数据库。从中下载并选取了多种长度的多肽片段,运用C'散点图的方法分析其结构特征。随机选择了FPA、TFPAV、CLV、GCLVK,用来说明 C'散点图这种方法在分析氨基酸结构中的意义。在FPA和TFPAV这两个多肽片段中,残基P的C'散点图如图2所示;在CLV和GCLVK这两个多肽片段中,残基L的C'散点图如图3所示。

图2 多肽片段FPA和TFPAV中残基P的C'散点图。(a)FPA;(b)TFPAVFig.2 The scatter plot of C'of residue‘P’from peptide fragments FPA and TFPAV.(a)FPA;(b)TFPAV
图2显示了残基P的骨架结构分别在FPA和TFPAV的分布情况。在图2中,可以非常直观地看到,残基P的 C'原子在片段 TFPAV中的分布要比在片段FPA中相对集中。在蛋白质中,片段FPA左右两边可以是任何氨基酸,TFPAV只是其中的一种情况。残基P在TFPAV中比在FPA中的序列相似度更高。所以,图2也印证了序列相似度越高,氨基酸结构相似度也越高,即序列决定结构。图3显示了残基L的骨架结构在多肽片段CLV和GCLVK的C'散点图。在图3中,也可以明显地看到GCLVK中的点要比 CLV集中。这表示,残基 L的结构在GCLVK中要比在 CLV中的更相似,与图2的结论是一致的。
3 讨论和结论
本研究提出的C'散点图,提供了一种描述多肽片段中氨基酸结构的新方法。与传统的二面角方法不同,C'散点图采用骨架原子C'相对于参考平面的坐标值来描述氨基酸结构的差异。由目标氨基酸的C'原子三维坐标描绘而成的散点图反映了这些氨基酸骨架的结构变化,图中每个点对应一个氨基酸的骨架结构。
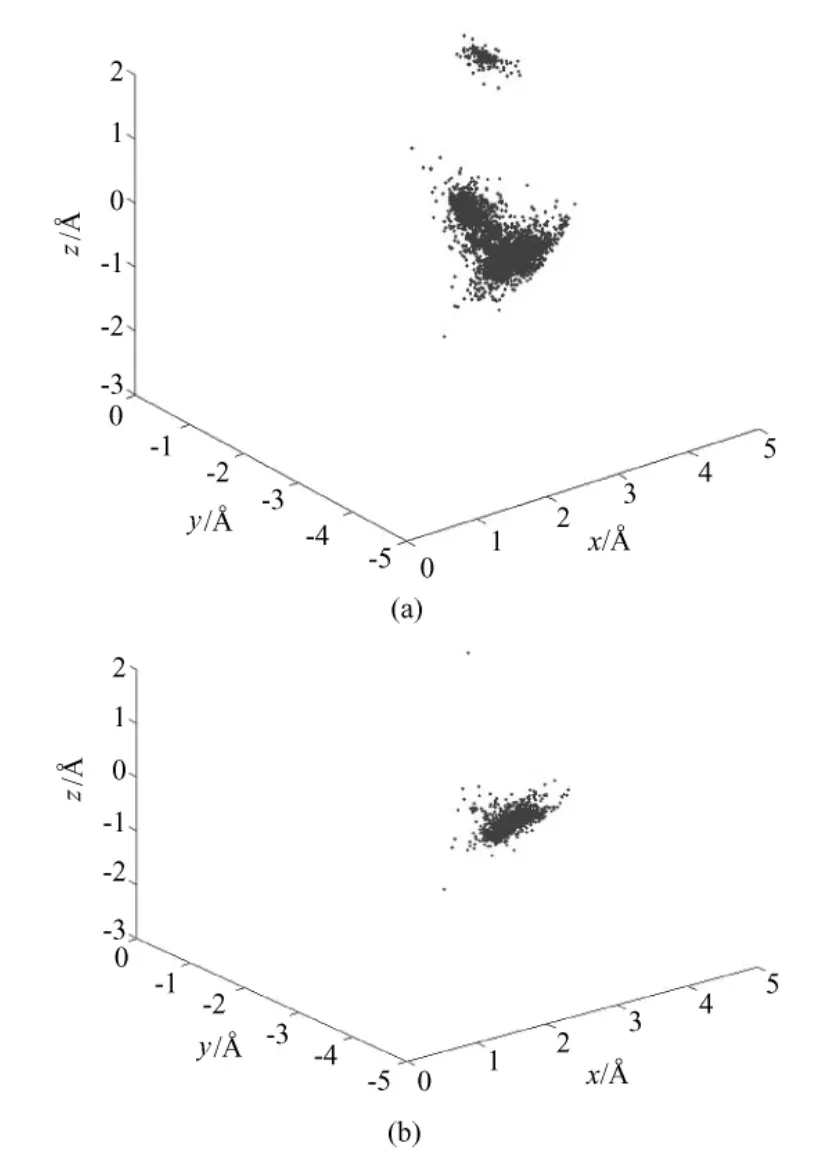
图3 多肽片段“CLV”和“GCLVK”中残基“L”的 C'散点图。(a)CLV;(b)GCLVKFig.3 The scatter plot of C'of residue‘L’from peptide fragments“CLV”and“GCLVK”.(a)CLV;(b)GCLVK
在运用C'散点图对氨基酸结构进行分析前,需要对原子坐标进行转换,使得C'原子的坐标值具有可比性,这比空间角的方法多一个步骤。不过整个转换过程并不复杂,所需的计算资源也不多,普通计算机就可以实现。
由于C'散点图是直接把原子的三维坐标显示在散点图中,所以这种方法比以往采用空间角的方法更加直观。同时,通过计算散点图中各点之间的距离,C'散点图还可以量化地评价氨基酸结构的多样性,使得用数字直接比较氨基酸结构的差异成为可能,这也是空间角的方法无法实现的。由于在散点图中存在一些相对分散的点,如图2(a),这些点的出现可能与蛋白质结构数据的误差有关。用两点间的最大距离计算氨基酸结构多样性时,这些点的出现会大大增加数值。在实际计算氨基酸结构多样性时如何处理这些点,是目前正在研究的问题。
图2和图3印证了氨基酸结构受序列相似度的影响。此外,从图中也可以看到,即使是5个残基相同的多肽片段,其中间的氨基酸结构还是有差异的。这说明影响氨基酸结构的因素是复杂的,局部的序列相同只能在一定程度上限制其结构的变化。今后,将运用C'散点图这一工具,进一步深入地研究影响氨基酸结构的因素,寻找氨基酸结构变化的规律。这些研究的结果,将对蛋白质结构预测的发展具有重要意义。
本研究提出了描述氨基酸结构的一种新方法——C'散点图。这种方法采用氨基酸骨架原子中C'的三维坐标值,反映对应氨基酸残基的结构变化。通过计算散点图上原子的分布情况,氨基酸结构的多样性也可以进行量化评价。C'散点图可以更加直观地分析氨基酸的结构特征和规律,对进一步深化氨基酸结构的分析和研究具有重要意义。
[1]Anfinsen C.Principles that govern the folding of protein chains[J].Science,1973,181:223 -230.
[2]Bystroff C,Simons KT,Han KF,et al.Local sequencestructure correlations in proteins[J].Curr Opin Biotechnol,1996,7:417-412.
[3]Crooks GE,Wolfe J,Brenner SE.Measurements of protein sequence-structure correlations[J]. Proteins StructFunct Bioinform,2004,57:804-810.
[4]Kabsch W,Sander C.On The use of sequence homologies to predict protein structure:identicalpentapeptidescan have completely different conformations[J].Proc Natl Acad Sci USA,1984,81:1075-1078.
[5]Pavlopoulou A,Michalopoulos I.State-of-the-art bioinformatics protein structure prediction tools[J].Int J Mol Med.2011.705.[Epub ahead of print]
[6]Ramachandran GN,Ramakrishnan C,Sasisekharan V,et al.Stereochemistry of polypeptide chain configurations[J].J Mol Biol,1963,7:95 -99.
[7]Pauling L,Corey RB,Branson HR.The structure of proteins;two hydrogen-bonded helical configurations of the polypeptide chain[J].Proc Natl Acad Sci USA,1951,37:205 -211.
[8]Esposito L, Vitagliano L, ZagariA, etal. Experimental evidence for the correlation of bond distances in peptide groups detected in ultrahigh-resolution protein structures[J].Protein Eng,2000,13:825-828.
[9]Malathy Sony SM,Saraboji K,Sukumar N,et al.Role of amino acid properties to determine backbone τ(N - Ca-C')stretching angle in peptides and proteins[J].Biophys Chem,2006,120:24-31.
[10]Gopalakrishnan K,Sowmiya G,Sheik SS,et al.Ramachandran plot on the web(2.0)[J].Protein Pept Lett,2007,14(7):669-671.
