不同晶型WO3·0.33H2O的结构及性能
2011-09-15胡栋虎贺蕴秋李林江季伶俐李一鸣
胡栋虎 贺蕴秋李林江 尹 婷 季伶俐 李一鸣
(同济大学材料科学与工程学院,上海 200092)
不同晶型WO3·0.33H2O的结构及性能
胡栋虎 贺蕴秋*李林江 尹 婷 季伶俐 李一鸣
(同济大学材料科学与工程学院,上海 200092)
以Na2WO4·2H2O为主要原料,采用液相法(80℃)和离子交换-水热法(150℃)分别制备了六方WO3·0.33H2O和以正交相为主的混合晶型WO3·0.33H2O。通过对2种晶型WO3·0.33H2O材料进行X射线衍射(XRD)、场发射电子扫描显微镜(FE-SEM)、红外光谱(FTIR)、X射线光电子能谱(XPS)和循环伏安测试,表征了产物的晶相和结构等。正交WO3·0.33H2O结构中由于相邻钨氧八面体层的相互位移而形成空隙,六方WO3·0.33H2O结构中没有位移则形成孔道;正交WO3·0.33H2O具有比六方WO3·0.33H2O更短键长的W=O和更负的导带位置。紫外-可见透射光谱研究表明,六方WO3·0.33H2O具有更明显的电致变色效应,可能是因为结构中的孔道使H+易扩散使六方WO3·0.33H2O更易发生氧化还原反应。光催化性能研究表明,正交WO3·0.33H2O具有更负的导带位置,价带电子跃迁后易于向电子受体转移,抑制了电子和空穴的复合,使得混合晶型WO3·0.33H2O的紫外光光催化能力相对六方WO3·0.33H2O更强。
六方WO3·0.33H2O;正交WO3·0.33H2O;电致变色;光催化
纳米半导体光催化材料具有高效率、低能耗及无二次污染等优点,在环境治理(尤其是污水处理)上备受关注[1-2]。目前除研究较多的TiO2[3-4]外,WO3体系材料由于其特有的光致变色[5-6]、电致变色[7-10]、气敏性[11-12]、湿敏性[13]和光催化[14-17]等性能,也越来越成为研究的热点。
WO3作为一种n型半导体材料,WO3的禁带宽度为 2.8 eV,与 TiO2(3.2 eV)相比,更具备光催化性能的潜力。但根据能带理论,只有当光催化剂的导带位置高于H+/H2氧化还原电位,且价带位置低于O2/H2O氧化还原电位时,材料才具有光催化性能。WO3的导带位置低于H+/H2氧化还原电位,因而不具备光催化性能。目前对WO3光催化性能的研究主要集中于对WO3进行离子掺杂[18-19]、半导体复合修饰[20-21]等,通过改变WO3的能带结构和抑制电子-空穴对的复合等来提高WO3系材料的光催化能力,而对WO3的结晶水合物(尤其是WO3·0.33H2O)光催化性能的研究却不多[22]。
实际上,关于WO3·0.33H2O的制备方法和结构已有不少研究和探索[22-31]。WO3·0.33H2O 存在两种晶型:正交 WO3·0.33H2O 和六方 WO3·0.33H2O。Zhou Liang 等[23]采用水热法制备出正交 WO3·0.33H2O,探讨了水热温度、H2O2的量和前驱物浓度等因素对产物物相和形貌的影响,同时对正交WO3·0.33H2O的结构进行了分析。Pfeifer等[24]通过衍射和红外等方法对正交WO3·0.33H2O的制备进行了详细的研究,指出Na的含量低于160 mg·kg-1时产物中含有WO3·H2O,高于 160 mg·kg-1时产物为正交 WO3·0.33H2O,Na 含量的增加有利于稳定正交 WO3·0.33H2O 晶相。
但目前很多关于WO3·0.33H2O的研究并未对六方 WO3·0.33H2O 和正交 WO3·0.33H2O 进行区分,及对二者结构和性能上的异同进行研究[22-31]。叶爱玲等[22]以钨粉和H2O2为反应物质,液相法制备出六方和正交混合晶型的WO3·0.33H2O纳米材料,发现该材料具有优异的紫外光光催化性能和良好的可见光光催化能力,同时具有光致变色效应,并对WO3、WO3·H2O 和 WO3·0.33H2O 的能带结构进行了研究,指出WO3·0.33H2O更负的导带位置是其具有优异光催化性能的原因。
本工作采用不同方法制备了六方WO3·0.33H2O及以正交为主要晶相的六方和正交混合晶型WO3·0.33H2O,着重研究两种不同晶型 WO3·0.33H2O 电致变色和光催化性能的差异,并通过FTIR、XPS和循环伏安等测试探讨导致这些性能差异的结构原因。
1 实验部分
1.1 材料的制备
使用的试剂均购自上海国药试剂有限公司,其中钨酸钠(Na2WO4·2H2O)和盐酸均为分析纯,阳离子交换树脂为732型 (Na型),溶剂为本实验室自制高纯水(ρ=17 MΩ·cm)。文中液相法制备的 WO3·0.33H2O 记为 H-WO3·0.33H2O,离子交换-水热法制备的 WO3·0.33H2O 记作 O-WO3·0.33H2O。
1.1.1 H-WO3·0.33H2O纳米粉体及其薄膜材料的制备
以Na2WO4·2H2O和盐酸为反应物质,在80℃水浴中反应1 h,生成淡黄色悬浊液H2WO4·nH2O(pH≈0.5),真空抽滤洗涤数次,80℃干燥后得到HWO3·0.33H2O纳米粉体。将抽滤后所得到的湿粉体用高纯水稀释,超声得到均匀的溶胶,然后在洁净ITO(Indium Tin Oxides,铟锡金属氧化物)玻璃上旋涂镀膜,在一定温度下烘干得到牢固的H-WO3·0.33H2O 薄膜材料。
1.1.2 O-WO3·0.33H2O纳米粉体及其薄膜材料的制备
实验以 0.2 mol·L-1Na2WO4溶液通过装有经处理的离子交换树脂的交换柱中,得黄色钨酸溶胶H2WO4·nH2O(pH≈1.5),将钨酸溶胶注入反应釜中,然后将洁净的ITO玻璃浸没于溶胶,150℃水热反应3 h,反应生成的白色悬浊液真空抽滤清洗数次,80℃干燥后得到O-WO3·0.33H2O纳米粉体,同时得到均匀牢固的O-WO3·0.33H2O薄膜材料。
六方和正交WO3·0.33H2O均由离子交换产物H2WO4·nH2O进一步反应而得,其化学反应可归纳如下:

1.2 样品表征
采用日本理学公司D/max-rB XRD分析仪测试样品的晶相和晶型,衍射源为铜靶(Cu Kα,λ=0.15418 nm),电流为60 mA,电压为40 kV,采用石墨单色器和闪烁计数器,扫描速度为10°·min-1,扫描范围为5°~70°;以UV-22501PC紫外可见吸收光谱仪测试六方 WO3·0.33H2O 和正交 WO3·0.33H2O 薄膜样品变色前后在紫外可见光波段的吸收谱图,测试范围为200~800 nm;采用Philips公司Quantum Esca型XPS仪测定六方WO3·0.33H2O和正交 WO3·0.33H2O材料中W4f的电子结合能,采用条件为铝/镁靶(14.0 kV,250 W);以 LK98BⅡ型电化学工作站测定薄膜样品的循环伏安曲线,并计算六方WO3·0.33H2O 和正交 WO3·0.33H2O 的氧化还原电位;样品分子结构表征采用德国布鲁克公司EQUINOX-55型红外光谱分析仪(FTIR),分辨率为 0.433 cm-1,测试范围为4000~400 cm-1;采用LS230型激光粒度分析仪(LPAS)研究粉体样品的颗粒粒径分布;采用S4800型场发射电子显微镜分析样品的微观形貌,电压为10 kV。
1.3 光催化性能测试
样品的光催化性能测试采用20 mg·L-1的甲基橙溶液作为目标降解物,取50 mg粉末样品与50 mL甲基橙溶液混合均匀,用2×125 W紫外光汞灯照射,每隔30 min取样一次,离洗数次后取上层清液测试透过率Ti(λ=483 nm),根据公式Ai=2-lgTi将Ti转换为吸光度Ai后代入以下公式计算甲基橙的降解率:
Di=(A0-Ai)/A0
(其中A0为光照前甲基橙在483 nm处的吸光度)
2 结果与讨论
2.1 XRD分析
图1所示为2种材料的XRD图。从图中可以看出,液相法和离子交换-水热法制备的材料均为WO3·0.33H2O(PDF No.350270、351001)。图 1(a)液相法制备的 WO3·0.33H2O的晶型为六方 WO3·0.33H2O(▲);图1(b)显示离子交换-水热法制备的材料在 2θ=18.02°、37.58°、51.92°、53.56°和 58.8°处出现正交 WO3·0.33H2O 的特征峰(◆),而 2θ=63.32°处的衍射峰则为六方WO3·0.33H2O的特征峰,除上述特征峰外,其余衍射峰的 2θ值(★)中,正交 WO3·0.33H2O和六方WO3·0.33H2O对应的2θ值极为相近,因此这些衍射峰应为六方WO3·0.33H2O和正交WO3·0.33H2O衍射峰的重叠,因此水热法制备的WO3·0.33H2O 是六方 WO3·0.33H2O 和正交 WO3·0.33H2O的混合晶相。也就是说,液相(常压)条件下形成纯六方WO3·0.33H2O晶相,而离子交换-水热法有利于生成正交WO3·0.33H2O。经计算O-WO3·0.33H2O 中正交 WO3·0.33H2O 与六方 WO3·0.33H2O的含量比例约为6.7∶1,即混合晶体中大多数为正交WO3·0.33H2O。实验发现,随着水热反应温度升高、时间延长和前驱物浓度增加等,O-WO3·0.33H2O中正交WO3·0.33H2O的含量逐渐增大,但仍含有部分六方WO3·0.33H2O,同时不利于产物性能的提高,论文给出的是光催化性能最强的O-WO3·0.33H2O。
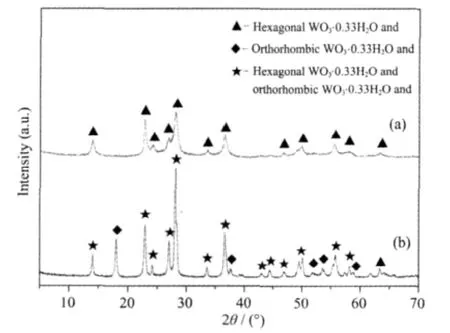
图1 H-WO3·0.33H2O(a)和 O-WO3·0.33H2O(b)的XRD图Fig.1 XRD patterns of H-WO3·0.33H2O(a)and O-WO3·0.33H2O(b)samples
WO3·0.33H2O晶体结构中含有2种钨氧八面体[23,30],一种只含有6个W-O单键,如图2(a),记作TypeⅠ;另一种除含有4个W-O单键外,其余2个氧原子分别与钨形成较短的W=O和较长的WOH2,即钨处于一个非对称的八面体中心,如图2(b),记作TypeⅡ;图2(c)和(d)中以四边形表示各层钨氧八面体的连接方式,实线箭头标记为TypeⅠ型钨氧八面体,虚线箭头表示TypeⅡ型钨氧八面体,2种钨氧八面体相互共顶连接形成钨氧八面体层,相邻钨氧八面体层间则通过TypeⅠ型共顶连接,而TypeⅡ中W=O和W-OH2不参与共顶。由PDF No.350270、351001可知,正交WO3·0.33H2O的晶格参数为:a=0.736 nm、b=1.251 nm、c=0.770 nm,而六方 WO3·0.33H2O为:a=0.729 nm、b=0.729 nm、c=0.388 nm,比较可得六方WO3·0.33H2O结构中晶格参数b和c约为正交 WO3·0.33H2O的1/2。比较图 2(c)和(d)可以看出,(c)中正交WO3·0.33H2O不同钨氧八面体层沿[010]方向相互位移b/2[22,29],而 (d)中六方WO3·0.33H2O在[010]方向上没有位移。这样六方WO3·0.33H2O在c轴方向上形成孔道,而正交WO3·0.33H2O由于位移没有孔道,只能形成空隙。
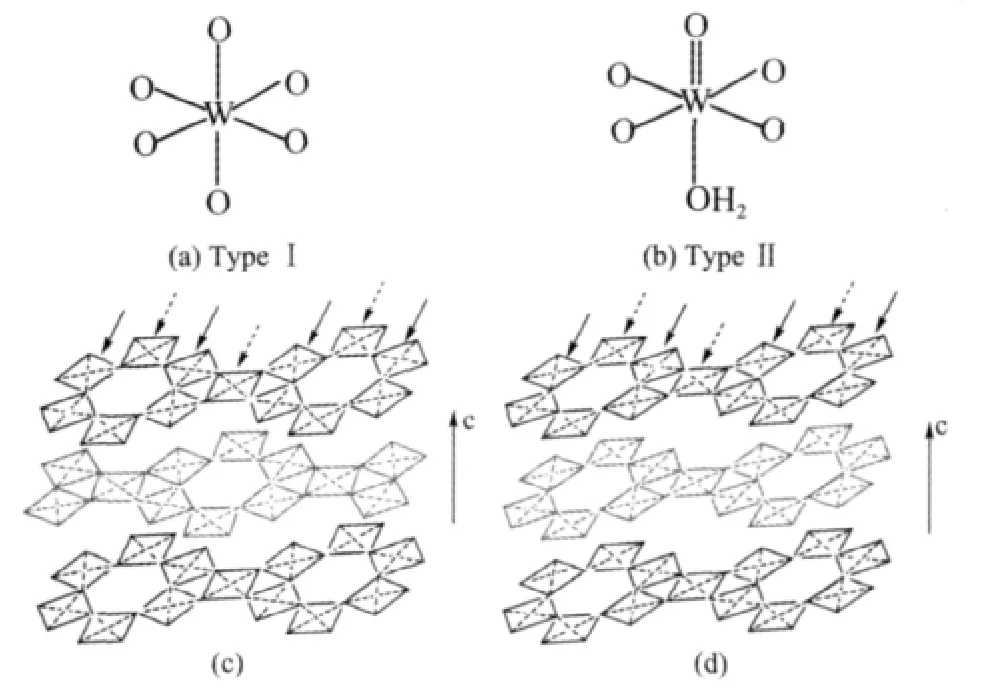
图2 正交 WO3·0.33H2O(c)和六方 WO3·0.33H2O(d)的结构Fig.2 Structure of orthorhombic WO3·0.33H2O(c)and hexagonal WO3·0.33H2O(d)samples
O-WO3·0.33H2O 和 H-WO3·0.33H2O 薄膜材料的制备条件与二者粉体材料相同。图3中扫描电镜照片显示 O-WO3·0.33H2O晶粒呈块状,H-WO3·0.33H2O中除杆状晶粒外,还含有更微小的粒状晶粒;O-WO3·0.33H2O 晶 粒的平均尺寸约为 50~100 nm,而H-WO3·0.33H2O中杆状晶粒约为25 nm。

2.2 FTIR分析
比较图4中O-WO3·0.33H2O 和 H-WO3·0.33H2O的红外光谱,可知二者在波数约为1 600 cm-1处存在O-H的弯曲振动峰,而缔合-OH特征峰分别在3494和3435 cm-1位置,由于二者均含有结构水,并可能含有吸附水,因此-OH的吸收峰可能是吸附水和结构水共同作用引起的;O-WO3·0.33H2O在1001 cm-1处有明显的W=O伸缩振动吸收峰,同时955 cm-1处的微弱吸收峰也归于O-WO3·0.33H2O中W=O的伸缩振动吸收峰,这与文献[30-31]中描述相吻合,而H-WO3·0.33H2O在W=O的振动波数范围没有出现明显的吸收峰,表明纯六方WO3·0.33H2O的红外光谱中没有W=O振动吸收峰,O-WO3·0.33H2O中 W=O振动吸收峰是由于正交 WO3·0.33H2O 引起的;从 O-WO3·0.33H2O 的红外光谱中可以看出,710和881 cm-1处均出现O-W-O伸缩振动吸收峰,而H-WO3·0.33H2O中O-W-O伸缩振动吸收峰出现在822 cm-1的位置。根据图2(c)和(d),正交WO3·0.33H2O的结构中相邻钨氧八面体层沿[010]方向相互位移 b/2,而六方 WO3·0.33H2O 没有位移。我们认为,对于六方 WO3·0.33H2O,上层钨氧八面体层中TypeⅡ型钨氧八面体的W=O与下层相邻钨氧八面体层中TypeⅡ型钨氧八面体的W-OH2之间可能形成氢键,导致W=O键长增大,使其在红外光谱中没有出现W=O振动吸收峰;而正交WO3·0.33H2O由于相邻层之间的相互位移,上层TypeⅡ型钨氧八面体的W=O和相邻下层中TypeⅡ型钨氧八面体的W-OH2之间不能形成氢键,使得W=O键长较六方WO3·0.33H2O短,这样在红外光谱中能观察到明显的W=O振动吸收峰。

图4 O-WO3.0.33H2O(a)和 H-WO3·0.33H2O(b)的红外光谱Fig.4 FTIR spectra of O-WO3·0.33H2O(a)and H-WO3.0.33H2O(b)samples
2.3 XPS分析
图5(a)、(b)分别为 H-WO3·0.33H2O 和 O-WO3·0.33H2O的W4f XPS 图谱。(a)中 H-WO3·0.33H2O 的W4f峰拟合后得到 35.4 eV的W4f7/2峰和 37.4 eV的 W4f5/2峰,(b)中 O-WO3·0.33H2O的W4f峰拟合后得到 35.5 eV的W4f7/2峰和 37.5 eV的W4f5/2峰,可以得出O-WO3·0.33H2O中W6+的电子结合能相对于H-WO3·0.33H2O 有所增加。由于 O-WO3·0.33H2O为以正交WO3·0.33H2O为主的混合晶体,而H-WO3·0.33H2O为纯六方 WO3·0.33H2O,表明 O-WO3·0.33H2O电子结合能的增加是正交WO3·0.33H2O引起的,可以推测正交WO3·0.33H2O的费米能级相对于六方WO3·0.33H2O更高。

图5 H-WO3·0.33H2O(a)和 O-WO3·0.33H2O(b)W4f XPS 图谱Fig.5 W4f XPS spectra of H-WO3·0.33H2O(a)and O-WO3·0.33H2O(b)
2.4 循环伏安分析
为探索 O-WO3·0.33H2O 和 H-WO3·0.33H2O 性能的差异,需要研究二者外层电子能带结构的差异,论文采用电化学工作站测量二者的循环伏安曲线,确定各自发生氧化还原反应的电位。实验以饱和甘汞电极为参比电极,表面镀有样品薄膜的ITO导电玻璃为工作电极,铂片为对电极,分别以浓度为0.5、1.0、2.0 mol·L-1的 HCl溶液作为电解质进行循环伏安测试,并设定电值流为正时属于还原电流。循环伏安曲线如图5所示。
图6中(a)、(c)、(e)分别是电解质溶液浓度为0.5、1.0 和 2.0 mol·L-1时 H-WO3·0.33H2O 的循环伏安曲线;(b)、(d)、(f)分别是溶液浓度为 0.5、1.0 和 2.0 mol·L-1时O-WO3·0.33H2O的循环伏安曲线。
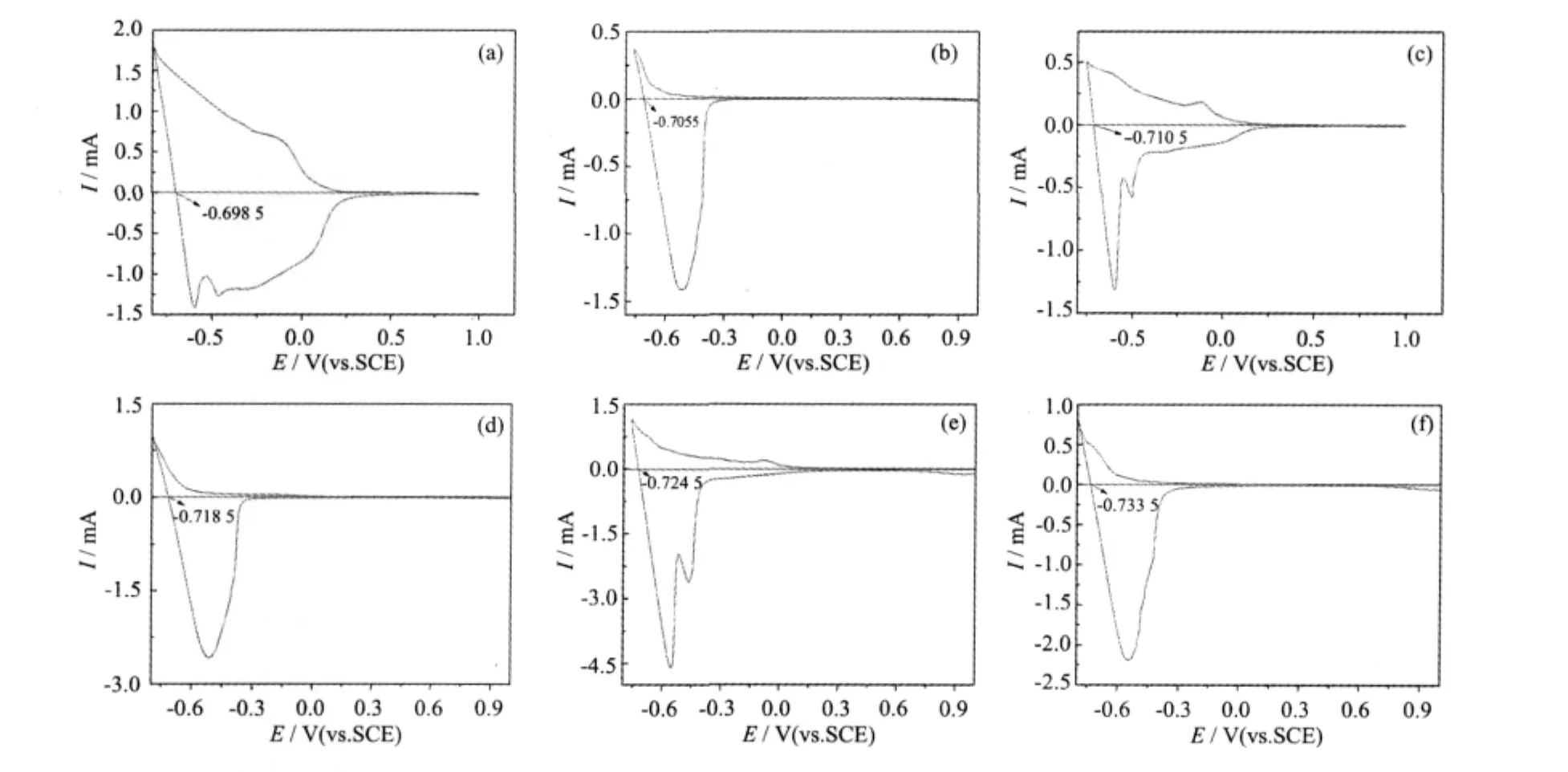
图6 不同电解质浓度时 H-WO3·0.33H2O(a、c、e)和O-WO3·0.33H2O(b、d、f)的循环伏安曲线(vs.SCE)Fig.6 Cyclic voltammograms of H-WO3·0.33H2O(a、c、e)and O-WO3·0.33H2O(b、d、f)in different concentration solution(vs.SCE)
图6中当电流为正时产生阴极电流,WO3·0.33H2O膜电极被还原;当电流为负时产生阳极电流,WO3·0.33H2O膜电极被氧化。产生阴极电流时WO3·0.33H2O膜电极上发生了如式(2)反应,电解质中H+进入WO3·0.33H2O结构,同时W6+获得电子被还原;产生阳极电流时膜电极上发生式(3)的逆反应,如式(3),W5+失去电子而被氧化。

从图6中可以看出,H-WO3·0.33H2O还原峰的面积比 O-WO3·0.33H2O 大;同时 H-WO3·0.33H2O 具有2个氧化峰,而O-WO3·0.33H2O只有一个氧化峰,这可能是因为H-WO3·0.33H2O中W6+还原为W5+后,部分 W5+继续还原为 W4+,而 O-WO3·0.33H2O 中W6+只得到了一个电子变为W5+。

图7 H-WO3·0.33H2O(a)和 O-WO3·0.33H2O(b)标准电位随溶液浓度变化曲线Fig.7 Standard potential of H-WO3·0.33H2O(a)and O-WO3·0.33H2O(b)as a function of concentration
零电流处为WO3·0.33H2O(vs.SCE)的平带电位,换算成标准平带电位如图7所示。由图7可知,HWO3·0.33H2O 和 O-WO3·0.33H2O 的平带电位均为负值,随着电解质溶液浓度的增大,二者的标准电位向更负方向变化;相同浓度时O-WO3·0.33H2O的标准电位较 H-WO3·0.33H2O 更负;O-WO3·0.33H2O 中主要为正交WO3·0.33H2O,意味着O-WO3·0.33H2O更负的平带电位是由正交WO3·0.33H2O引起的。我们认为,可能由于六方WO3·0.33H2O在c轴方向上的孔道,有利于H+的扩散及六方WO3·0.33H2O的氧化还原反应,而正交WO3·0.33H2O结构中只能形成空隙,不利于H+的进入,因此正交WO3·0.33H2O发生氧化还原反应所需能量更大。
由此可知,正交WO3·0.33H2O和六方WO3·0.33H2O均具有比H+/H2能级更负的导带电位,并且正交 WO3·0.33H2O的导带电位比六方 WO3·0.33H2O更负。
2.5 电致变色性能
在循环伏安测试中我们发现,H-WO3·0.33H2O膜电极被还原时由无色变为深蓝色,而后被氧化时又逐渐变为无色;相应地,O-WO3·0.33H2O膜电极先由无色变为淡红棕色,再逐渐退色至无色。
实验取电流达到正的最大值,WO3·0.33H2O膜电极颜色变化最深时的样品,测试其紫外-可见光的吸收情况,如图8所示。实验发现,H-WO3·0.33H2O膜变色前后对紫外-可见光吸收性能有明显变化。电致变色前,H-WO3·0.33H2O几乎不吸收可见光,而在变色后材料对可见光的吸收有显著增强。但是O-WO3·0.33H2O膜变色前后对紫外-可见光的吸收也没有明显的变化。两种晶型电致变色性能的差异与二者的循环伏安性能是对应的,均可能源于前述分析中六方 WO3·0.33H2O和正交 WO3·0.33H2O的结构差异。
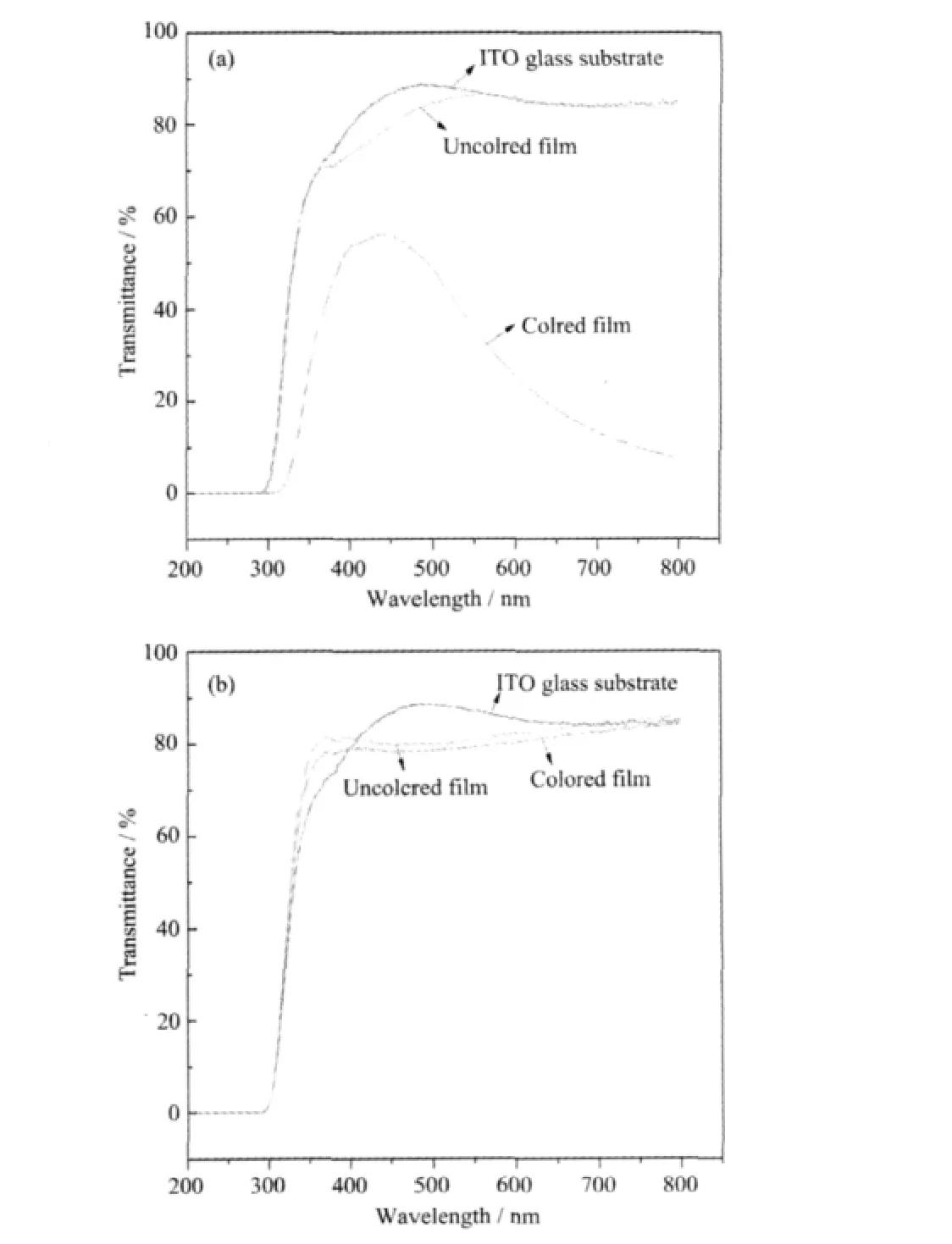
图8 H-WO3·0.33H2O(a)和 O-WO3·0.33H2O(b)变色前后的紫外-可见透射光谱Fig.8 UV-Vis transmittance spectra of H-WO3·0.33H2O(a)and O-WO3·0.33H2O(b)films
2.6 光催化性能
图9中二者光催化结果表明O-WO3·0.33H2O在紫外光照条件下对甲基橙的降解率达到58%,而H-WO3·0.33H2O 只 有 39%。图 10为 H-WO3·0.33H2O和O-WO3·0.33H2O粉体材料的颗粒粒径分布曲线,结果表明,两种不同条件下制备的WO3·0.33H2O材料的平均颗粒粒径均约为130 nm。与扫描电镜分析相比,O-WO3·0.33H2O的晶粒尺寸与其粉体材料颗粒粒径分布基本一致,H-WO3·0.33H2O的晶粒尺寸远小于粉体材料颗粒粒径分布,这可能是由于团聚引起的。可以推测,二者光催化性能上的差异是由于材料本身结构的不同引起的,而OWO3·0.33H2O较高的光催化性能应归因于正交WO3·0.33H2O。由于正交 WO3·0.33H2O 比六方 WO3·0.33H2O具有键长更短的W=O,而XPS和循环伏安分析表明,正交WO3·0.33H2O具有更高的费米能级和导带位置,价带上电子跃迁后,在导带和价带上形成电子-空穴对,价带上的空穴具有光催化作用,正交WO3·0.33H2O更负的导带位置更易于电子向H+等介质的转移,从而利于抑制电子与空穴的复合,使价带上参与光催化作用的空穴相对较多,使其具有相对较高的光催化性能。

图9 O-WO3·0.33H2O(a)和 H-WO3·0.33H2O(b)的紫外光光催化性能曲线Fig.9 Photocatalytic degradation curves of O-WO3·0.33H2O(a)and H-WO3·0.33H2O(b)samples under UV-light irradiation

图10 H-WO3·0.33H2O(a)和 O-WO3·0.33H2O(b)样品的颗粒粒径分布曲线Fig.10 Particle size distribution curves of H-WO3·0.33H2O(a)and O-WO3·0.33H2O(b)samples
3 结 论
研究了采用不同方法制备的纯六方WO3·0.33H2O和以正交相为主的正交-六方混合的WO3·0.33H2O的结构和性能。
六方WO3·0.33H2O结构中的孔道,易于H+的扩散,有利于六方WO3·0.33H2O的氧化还原反应,从而具有明显的电致变色效应;而正交WO3·0.33H2O结构中无连通孔道,不利于H+的进入,使以正交相为主的WO3·0.33H2O电致变色性能不明显。
正交WO3·0.33H2O的结构中相邻钨氧八面体层沿[010]方向位移b/2,上层中的W=O和相邻下层中W-OH2之间不能形成氢键,而六方WO3·0.33H2O结构中没有位移,层间可以形成氢键。这一结构差异使正交WO3·0.33H2O保持了键长较短的W=O,其费米能级和导带位置比六方WO3·0.33H2O更高,因此价带上电子跃迁后更易于转移,有利于抑制电子和空穴的复合,使得以正交相为主的混合晶体WO3·0.33H2O的紫外光光催化活性更高。
[1]Eggins B R,Palmer F L,Byrne J A,et al.Water Res.,1997,31(5):1223-1226
[2]Ochuma I J,Osibo O O,Fishwick R P,et al.Catal.Today,2007,128:100-107
[3]Zhang W J,He Y Q,Qi Q,et al.Mater.Chem.Phys.,2005,93:508-515
[4]Li D,Haneda H,Labhsetwar N K,et al.Chem.Phys.Lett.,2005,401(4/5/6):579-584
[5]Bechinger C,Oefinger G,Herminghaus S,et al.J.Appl.Phys.,1993,74(7):4527-4533
[6]Xu N,Sun M,Cao Y W,et al.Appl.Surf.Sci.,2000,157(1/2):81-84
[7]Davazoglou D,Donnadieu A.Thin Solid Films,1998,164:369-374
[8]Krasnov Y S,Kolbasov G Y.Electrochimica.Acta,2004,49(15):2425-2433
[9]Sallard S,Brezesinski T,Smarsly B M.J.Phys.Chem.C,2007,111:7200-~7206
[10]YE Hui(叶辉),LIU Xiao-Yan(刘晓艳).Chinese J.Acta Optica Sinica(Guangxue Xuebao),1999,19(4):532-539
[11]WEI Shao-Hong(魏少红),NIU Xiao-Yu(牛晓玉),CHENG Zhan-Sheng(成 战 胜).Chinese J.Electron.Components Mater.,2004,23(10):12-16
[12]Antonik M D,Schneider J E,Wittman E L,et al.Thin Solid Films,1995,256(1/2):247-252
[13]Parvatikar N,Jain S,Khasim S,et al.Sens.Actuators,B:Chem.,2006,114(2):559-603
[14]Bamwenda G R,Arakawa H.Appl.Catal.A:General,2001,201:181-191
[15]Wang H Y,Xu P,Wang T M.Mater.Des.,2002,23:331-336
[16]ZOU Li-Xia(邹丽霞),ZHONG Qing(钟秦),LIU Qing-Cheng(刘庆成),et al.Chinese J.Chem.Ind.Eng.Prog.(Huagong Jinzhan),2005,24(9):1015-1019
[17]Yang B,Barnes P R F,Zhang Y J.Catal.Lett.,2007,118:280-284
[18]Cheng X F,Leng W H,Liu D P,et al.Chemosphere,2007,68:1976-1984
[19]Porkodi P,Yegnaraman V,Jeyakumar D,et al.Mater.Res.Bull.,2006,41:1476-1486
[20]Kwon Y T,Song K Y,Lee W I,et al.J.Catal.,2000,191:192-199
[21]CHENG Ying-Zhi(成英之),ZHANG Yuan-Ming(张渊明),TANGYu(唐渝).Chinese J.Catal.(Cuihua Xuebao),2001,22(2):203-205
[22]YE Ai-Ling(叶爱玲),HE Yun-Qiu(贺蕴秋),ZHOU Wen-Ming(周文明),et al.Chinese J.Funct.Mater.(Gongneng Cailiao),2009,40(1):52-59
[23]Zhou L,Zou J,Yu M M,et al.Cryst.Grouth Des.,2008,8(11):3993-3998
[24]Pfeifer J,Cao G F,Tekula-Buxbaum P,et al.J.Solid State Chem.,1995,119:90-97
[25]Gerand B,Nowogrocki G,Figlarz M.J.Solid State Chem.,1981,38:312-320
[26]Gotic M,Ivanda M,Popovic S,et al.Mater.Sci.Eng.,B,2000,77:193-201
[27]Pecquenard B,Lecacheux H,Livage J,et al.J.Solid State Chem.,1998,135:159-168
[28]Nogueira H I S,Cavaleiro A M V,Rocha J,et al.Mater.Res.Bull.,2004,39:683-693
[29]Balazsi C,Pfeifer J.Solid State Ionics,2002,151:353-358
[30]Guéry C,Choquet C,Dujeancourt F,et al.J.Solid State Electrochem,2007,1:199-207
[31]Daniel M F,Desbat B,Lasseegues J C.J.Solid State Chem.,1987,67:235-247
Structure and Properties of WO3·0.33H2O Polymorphs
HU Dong-Hu HE Yun-Qiu* LI Lin-Jiang YIN Ting JI Ling-LiLI Yi-Ming
(School of Material Science and Engineering,Tongji University,Shanghai 200092,China)
Hexagonal WO3·0.33H2O and mixed WO3·0.33H2O(orthorhombic WO3·0.33H2O as the main phase)were synthesized by liquid method (80℃)and by ion-exchange and hydrothermal process(150℃),respectively,using Na2WO4·2H2O as the precursor.The samples were characterized by X-ray diffraction (XRD),Field Emission Scanning Electron Microscopy(FE-SEM),infrared spectra (FTIR),X-ray Photoelectron Spectroscopy (XPS)and Cyclic voltommetry.The inter-shifting of adjacent WO6octahedron layers in orthorhombic WO3·0.33H2O produces voids,but ways appear in hexagonal WO3·0.33H2O for no displacement;Orthorhombic WO3·0.33H2O had shorter W=O and more minus conduction band than hexagonal WO3·0.33H2O.UV-Vis transmittance spectra indicate that more distinct electrochromism of hexagonal WO3·0.33H2O may be responsible for easy H+diffusion into the structure and favorable to redox reaction.Photocatalytic property of mixed WO3·0.33H2O is better than hexagonal WO3·0.33H2O,because more minus conduction bands of orthorhombic WO3·0.33H2O are brought about due to easy absorption of electrons from valence band by electron acceptors and recombination restriction of electron-holes.
hexagonal WO3·0.33H2O;orthorhombic WO3·0.33H2O;electrochromism;photocatalytic property
O643.61+3;TQ132.4+1
A
1001-4861(2011)01-0011-08
2010-07-20。收修改稿日期:2010-09-19。
国家自然科学基金(No.50672066)资助项目。
*通讯联系人。E-mail:heyunqiu@mail.tongji.edu.cn,Tel:021-69582117
