高效液相色谱法测定舒冠胶囊中淫羊藿苷含量
2011-07-28万维香
万维香
(江苏省泰州市第一人民医院,江苏 泰州 225300)
舒冠胶囊由制何首乌、川芎、黄精(制)、红花、淫羊藿、五灵脂(醋制)、丹参7味药材的提取物加适宜的辅料制成,制何首乌为方中主药,含有大黄素、大黄素甲醚、大黄酚等蒽醌类脂溶性成分和2,3,5,4'-四羟基二苯乙烯-2-O-β-D-葡萄糖苷等二苯乙烯类水溶性等成分[1]。由于大黄素、大黄素甲醚等在水提醇沉工艺中转移率较低,而2,3,5,4'-四羟基二苯乙烯-2-O-β-D-葡萄糖苷遇光极不稳定,此两类成分不宜作为含量控制指标。因此笔者参考文献[2],选择淫羊藿的主要成分淫羊藿苷作为指标成分,采用准确、灵敏的高效液相色谱法进行含量测定,现报道如下。
1 仪器与试药
Model 510型液相色谱仪(美国Waters);TG332A型电子天平(湘西仪器仪表厂)。淫羊藿苷对照品(中国药品生物制品检定所,批号为0737-200111,供含量测定用);舒冠胶囊(自制,批号分别为041206,041207,041208);所用甲醇为色谱纯,其余试药与溶剂均为分析纯;水为重蒸水。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:用十八烷基硅烷键合硅胶为填充剂的Shimpack-ODS柱(150 mm×4.5 mm,5 μm);检测波长:270 nm;流动相:甲醇-0.05%磷酸溶液(55∶45);流速:1 mL/min。在此色谱条件下,色谱图见图1,淫羊藿苷峰理论板数为2518,拖尾因子 T=1.106,分离度R=1.94,符合规定。

图1 高效液相色谱图
2.2 溶液制备
取本品内容物约0.2 g,精密称定,置具塞三角烧瓶中,精密加入流动相50 mL,称定质量,加热使溶解,放冷,再称定质量,用流动相补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。按照处方组成,取除淫羊藿以外的其余辅料,按制备工艺要求制成不含淫羊藿舒冠胶囊,按供试品溶液制备项下方法制备成阴性对照品溶液。取淫羊藿苷对照品适量,精密称定,加流动相制成每1 mL含25μg的对照品溶液。
2.3 方法学考察
阴性干扰试验:照拟订的色谱条件,精密吸取阴性对照品溶液20 μL,注入液相色谱仪,记录色谱图。从图1可知,阴性对照品溶液在淫羊藿苷峰位置无吸收峰,说明阴性对照品溶液不干扰主药的含量测定。
线性关系考察:取淫羊藿苷对照品,加流动相制成47.6μg/mL的溶液,作为贮备液。分别精密吸取贮备液,加流动相制成质量浓度为 4.76,9.52,19.04,28.56,38.08,47.6μg/mL 的溶液,按上述色谱条件测定峰面积。以峰面积为纵坐标(Y)、淫羊藿苷进样量为横坐标(X)进行回归处理,得线性回归方程 Y=2000000X+60496,r=0.9999。结果表明,淫羊藿苷进样量在 0.0952 ~ 0.952μg 范围内与峰面积呈良好的线性关系。
精密度试验:取对照品溶液,精密吸取20 μL,重复进样6次。结果的 RSD=0.97%(n=6),表明方法精密度良好。
重现性试验:取同一批样品6份,依法制备供试品溶液并测定淫羊藿苷含量,结果的 RSD=2.0%(n=6),表明方法重现性良好。
稳定性试验:取同一供试品溶液,于室温条件下放置8 h,每隔2 h进样1次,测定峰面积。结果的 RSD=1.7%(n=5),表明供试品溶液在8 h内稳定。
加样回收试验:取已知含量的样品(含量2.8 mg/粒)6份,每份约0.1 g,置具塞三角烧瓶中,分别精密加入淫羊藿苷对照品10 mL(55μg/mL,流动相溶解),再精密加入流动相 40 mL,称定质量,加热使溶解,放冷,再称定质量,用流动相补足减失的质量,摇匀,滤过,取续滤液,依法测定,结果见表1。
2.4 样品含量测定
分别精密量取10批样品适量,按溶液制备方法制备对照品溶液和供试品溶液,精密吸取对照品溶液与供试品溶液各20 μL,注入液相色谱仪,依法测定。结果见表2。
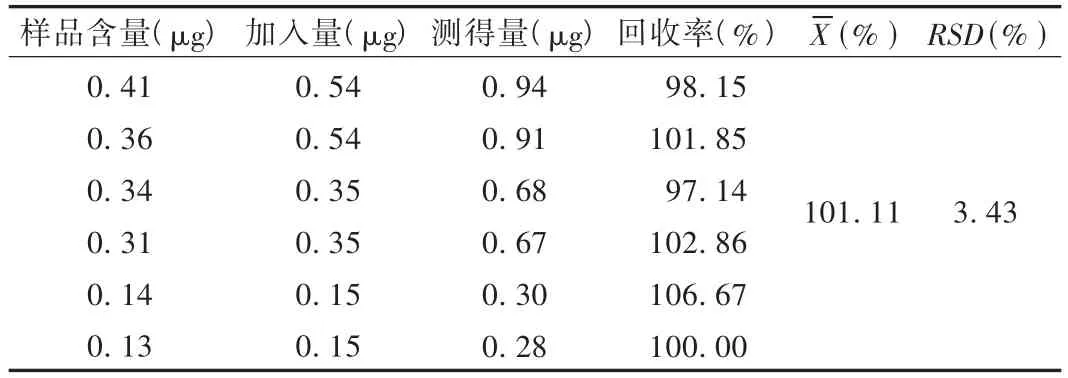
表1 加样回收试验结果(n=6)
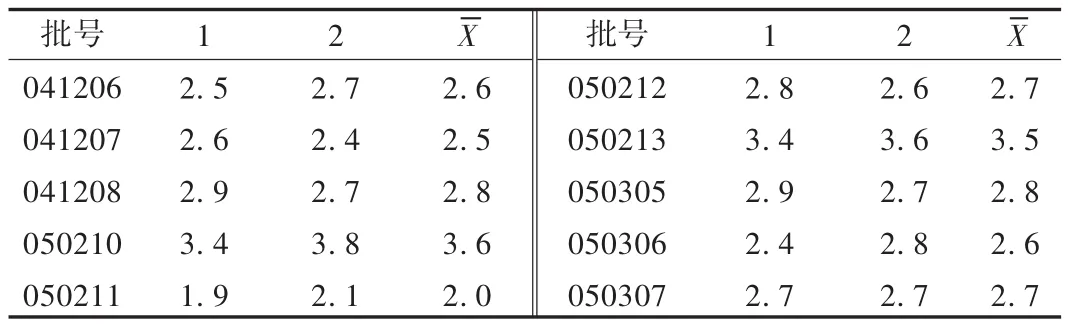
表2 样品含量测定结果(mg/粒,n=10)
2000年版《中国药典(一部)》[3]规定淫羊藿药材含淫羊藿苷(C33H40O15)不少于0.5%,本品每粒含淫羊藿药材0.6 g,理论上本品每粒含淫羊藿苷应不低于3 mg。实测本品10批样品,淫羊藿苷含量为2.0~3.6 mg/粒;稳定性试验中,含量基本稳定。考虑到生产上的波动性,故暂订本品淫羊藿苷含量不低于1.8 mg/粒[4-5]。
3 讨论
取淫羊藿苷对照品适量,加流动相制成每1 mL含10μg的溶液,在250~300 nm波长范围内扫描,结果本品在269 nm波长处有最大吸收,参考中国药典淫羊藿苷的检测波长270 nm,故确定270 nm为检测波长。
比较了 53 ∶47,56∶44,55∶45不同配比的甲醇-0.05%磷酸溶液作为流动相,结果以55∶45所得色谱图较优,基线回落良好。
方法学考察结果表明,所建立的含量测定方法简单、结果准确、专属性强,能控制产品质量。
[1]郭艳玲,王玉珍,陈再兴,等.高效液相色谱法测定舒冠胶囊中丹参酮ⅡA含量[J].中药材,2004,27(5):379-380.
[2]梁生旺,王淑美,冯素香,等.RP-HPLC测定冠心舒胶囊中丹酚酸B及芍药苷的含量[J].中成药,2007,29(3):462-465.
[3]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2000:122,229,268.
[4]权迎春,郑光浩,关丽萍.延边大学医学学报[J].延边大学医学学报,2007,30(2):108-110.
