HPLC法和聚类分析法探讨不同厂家消癌平片中绿原酸含量的一致性和稳定性
2011-07-25温瑞卿李东辉
温瑞卿, 李东辉, 王 鑫
(北京市海淀区药品检验所,北京100083)
消癌平片具有抗癌、消炎、平喘作用,主要用于治疗食道癌、胃癌等多种恶性肿瘤[1-4],是当前为数不多的抗癌中成药之一。该制剂由药材乌骨藤(通关藤)经水煮、浓缩、加辅料压片而成,虽然原料和工艺均比较单一,但其原料药质量参差不齐[5-11],且各厂家质量标准(见各厂家的散页标准)规定项目、方法和限度不同[12],因此各厂家批次质量的一致性无法得到有效控制。乌骨藤抗癌的药效成分主要有酯苷、酚酸和多糖三大类,其中酯苷类成分含量低、且无对照品对照和标定,因此目前主要针对绿原酸类成分定性和定量控制制剂质量[13-15],本研究即探讨北京地区流通和使用的消癌平片中绿原酸含量差异,旨在为考察制剂质量的一致性和稳定性提供参考,为进一步制定和完善消癌平片的质量标准提供数据支持。
1 仪器与试药
Waters2695高效液相色谱仪,Waters2998 PDA检测器,AS10200BT超声波提取仪,Sartorius A200S电子天平,Sartorius BP211D电子天平,ACY-1002-U超纯水机。
消癌平片,绿原酸对照品(中国药品生物制品检定所提供,批号:110753-200413)。
乙腈、磷酸均为色谱纯,水(实验室自制超纯水,电阻率18.25 mΩ.cm)。
2 方法与结果
2.1 色谱条件 色谱柱:Kromasil-C18色谱柱(4.6 mm×150 mm,5 μm);流动相:乙腈 -0.4% 磷酸溶液(9 ︰ 91);检测波长:327 nm;柱温:30℃;体积流量:1.0 mL/min;进样量10 μL或20 μL。在此色谱条件下,绿原酸与其他峰可彻底分离,分离度R≥1.5,理论塔板数>2000。色谱图见图1。

图1 对照品(A)及供试品(B)色谱图
2.2 对照品溶液的制备 精密称取绿原酸对照品9.32 mg,置50 mL棕色量瓶中,用50%甲醇溶解并稀释至刻度,摇匀,精密吸取2 mL,置20 mL棕色量瓶中,用50%甲醇稀释至刻度,摇匀,即得(每1 mL中含18.64 μg绿原酸)。
2.3 供试品溶液的制备 取消癌平片20片,除去包衣,精密称定,研细,取0.6 g,置具塞锥形瓶中,精密加50%甲醇25 mL,称定质量,超声30 min(功率200 W,频率40 KHz),放冷,称定质量,用50%甲醇补足减失的质量,摇匀,用微孔滤膜(0.45 μm)滤过,即得。
2.4 线性关系考察 精密吸取上述绿原酸对照品溶液4、8、12、16、20、24 μL,分别注入液相色谱仪,按上述色谱条件测定峰面积,以进样量为横坐标(X),以峰面积积分值为纵坐标(Y),进行回归处理,结果绿原酸在0.07456~0.4474 μg范围内,进样量与峰面积积分值之间线性关系良好,回归方程为 Y=3 ×106X+42128,r=0.9997。
2.5 精密度试验 精密吸取对照品溶液12 μL,连续进样5次,测定绿原酸峰面积,结果平均峰面积为6.896×105,相对标准偏差RSD为1.1%。
2.6 稳定性试验 精密吸取同一供试品溶液分别于0、1、2、7、10、15、18 h 进样,结果绿原酸峰面积的相对标准偏差RSD 为0.5%。
2.7 重复性试验 取同一样品,平行制备5份供试品溶液,按上述条件测定,结果绿原酸含量均为0.13 mg/片,相对标准偏差RSD为0%。
2.8 加样回收率试验 取重复性试验项下的样品5份,分别定量加入绿原酸对照品溶液(18.64 μg/mL)1 mL,挥干,同法提取并测定,计算回收率,分别为96.4%、97.5%、95.0%、98.2%、96.7%,平均回收率为 97%,相对标准偏差RSD 为1.3%。
2.9 样品测定 取8个厂家共20批次样品,按上述方法测定,将峰面积代入线性方程,计算绿原酸量,结果见表1:
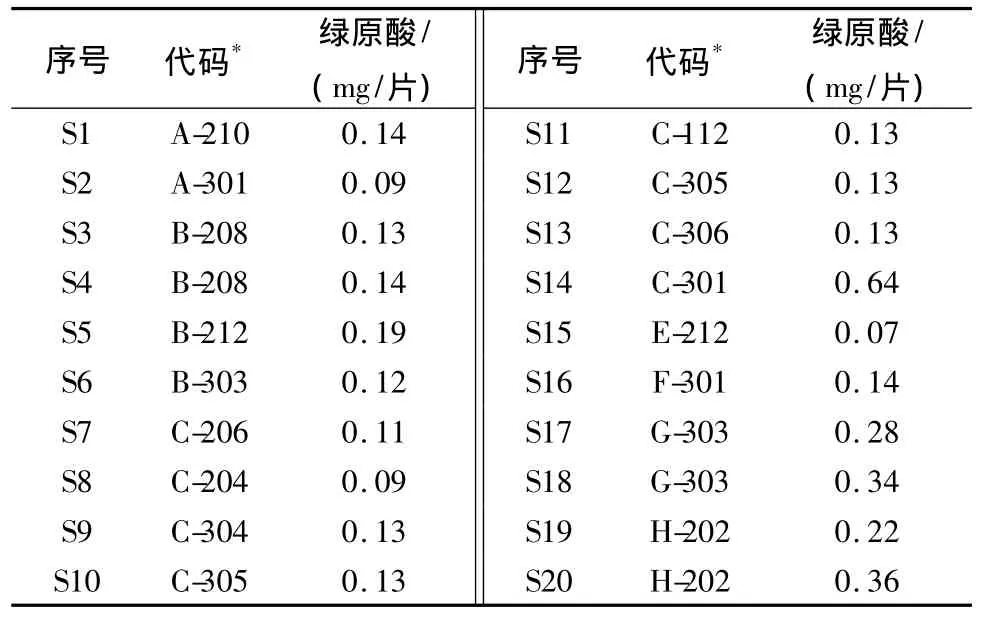
表1 不同厂家批次样品中绿原酸测定结果
3 各厂家批次样品的比较
3.1 各厂家批次样品的比较 将表1中20批次消癌平片的绿原酸含量输入SPSS16.0软件,采用组间均连法,以欧氏距离为指标进行聚类分析,结果如图2所示,A、B、C、E、F厂家的14批次样品的绿原酸含量非常接近,与G厂家S17、H厂家S19以及B厂家S5比较接近;G厂家S18、H厂家S20非常接近;D厂家S14绿原酸含量最高,独立成一类。
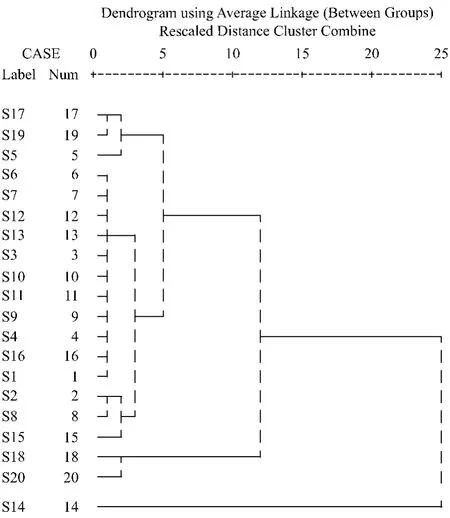
图2 各批次样品绿原酸含量的聚类分析图
3.2 稳定性分析 S3与 S4、S10与 S12、S17与 S18、S19与S20均为同厂家同批号样品,其中第一组内2批次样品从同一使用单位抽样,后3组中2批次样品从不同经营或使用单位抽样,但前2组内2批次样品的绿原酸含量相近或相同,后2组内2批次样品差异很大,聚类分析将其交叉归类,即S17和S19、S18和S20分别属于两类。这可能由于绿原酸的稳定性较差,某些样品因运输和贮存条件控制不严而导致绿原酸含量降低。
综合一致性和稳定性结果,按照绿原酸含量由高至低,大致可将上述厂家样品划为3个水平,即D>H-G>B-F-AC-E。
4讨论
各厂家纷纷申请了各自的散页标准,但规定的检测项目、方法和含量限度均不一致,导致不同厂家药品的质量很难保持一致。本文在执行各厂家标准进行法定检验的基础上,摸索了绿原酸和咖啡酸的含量测定方法,在文中所述条件下可同时测定2种成分(图1),但因咖啡酸为绿原酸分解后产物,制剂中含量很低(5~10 μg/片),对于控制和比较制剂质量的意义很小,故本文仅采用聚类分析法比较了各厂家批次样品绿原酸含量的一致性和稳定性,希望为考察制剂质量的一致性和稳定性提供参考,为制定和完善消癌平片的质量标准提供数据支持。
因中国食品药品检定研究院目前只能提供绿原酸和咖啡酸对照品,所以本次研究的指标单一,无法全面反映消癌平片药效物质基础,接下来我们将采用HPLC特征图谱更全面比较各样品的质量差异,进一步为评价制剂质量和完善质量标准提供依据。
[1]朱萱萱,赵陆华,严士海,等.乌骨藤提取物对人胃癌细胞(MKN28)细胞增殖的研究[J].实用中医内科杂志,2007,21(6):36-39.
[2]赵陆华,朱萱萱,张忠华,等.乌骨藤提取物对人肝癌细胞(HepG2)增殖的实验研究[J].中华中医药学刊,2008,26(12):2633-2635.
[3]陈同生,王小平,王治平,等.消癌平诱导人肺腺癌(ASTC-a-1)细胞内caspase-3活化的荧光光谱分析[J].光谱学与光谱分析,2008,28(6):1327-1331.
[4]刘长余.通关藤及其制剂的临床应用[J].海峡药学,2006,18(5):156-157.
[5]才 凤,张 慧,初正云.通关藤及其伪品的FTIR鉴别[J].辽宁中医药大学学报,2007,9(4):58-59.
[6]赖玉菡,王 曙.HPLC测定通关藤中绿原酸的含量[J].华西药学杂志,2006,21(1):079-081.
[7]陈 强,毛春芹,陆兔林,等.不同产地通关藤药材中总皂苷与通关藤苷 B的含量比较[J].医药导报,2009,28(12):1615-1617.
[8]赵陆华,相秉仁,陆红柳.乌骨藤药材HPLC指纹图谱研究[J].中成药,2008,30(8):1093-1096.
[9]马 尧,赵陆华,肖望书,等.HPLC-UV法测定通光藤药材中通光藤皂苷 A[J].中草药,2009,40(5):820-821.
[10]何 俊,王 曙,严晓梁.不同产地的通光藤中总皂苷含量的比较[J].华西药学杂志,2008,23(2):201-202.
[11]王月敏,夏素霞,于绍军,等.乌骨藤药材中绿原酸的含量测定方法研究[J].中国实验方剂学杂志,2007,13(7):12-13.
[12]卫生部药品标准中药成方制剂第二十册[S].1998:277.
[13]谭朝阳,雷玉萍.HPLC法测定乌骨藤及其制剂消癌平片中绿原酸的含量研究[J].中医药学刊,2004,22(7):1354.
[14]于文静,王焕群.HPLC法测定消癌平口服液中绿原酸的含量[J].中国药品标准,2006,7(3):50-51,76.
[15]赵陆华,相秉仁,谭喜莹.高效毛细管电泳法测定乌骨藤药材及其制剂中绿原酸的含量[J].中国现代应用药学杂志,2008,25(2):152-154.
