Rho激酶抑制剂短期治疗射血分数保留性心衰疗效评估
2011-06-18马小川高智耀
张 伟,马小川*,高智耀*,王 芳,张 涛
30%~50%的心衰患者,在拥有心衰症状的同时,仍能维持正常的左室射血分数。2008年,欧洲心脏病学会(ESC)的心衰指南中,建议将其称为左室射血分数保留性心力衰竭(Heart failure with preserved left ventricular ejection fraction,HFPEF)。HFPEF为一种复杂的临床综合征,其发病机制至今不明确。ACEI/ARB类药物、醛固酮拮抗剂和正性肌力药物等,在最佳药物治疗(Optimal medical therapy,OMT)方案的基础上,并不能进一步改善其主要不良终点事件,提示还有不同的分子生物学机制参与了HFPEF的病理过程。
近年研究发现,小分子GTP酶家族成员Rho激酶与其天然配体Rho蛋白A结合,在多种细胞生长因子的调节下,能促进细胞内应力纤维及黏着斑的形成,从而对细胞骨架重排具有调控作用,进而参与调节细胞的局部黏附、迁移、聚集、增殖和基因转录等生物学功能[1-2]。在心血管研究领域,Rho激酶抑制剂通过抑制肌球蛋白轻链磷酸化而抑制血管平滑肌收缩,从而达到扩张血管、调节血压、改善循环状态的作用;通过抑制局部心肌组织炎性细胞分泌各种炎症介质因子,降低炎症损伤程度,如Rho激酶抑制剂法舒地尔对动脉粥样硬化中单核细胞趋化蛋白-1(MCP-1)的抑制作用[3];对血管紧张素 II(AngiotensinⅡ,Ang II)诱导的心肌纤维化及重构具有干预作用[4]。本研究观察Rho激酶抑制剂法舒地尔短期治疗对HFPEF患者心脏舒张功能指标的干预作用,旨在明确其在此类心衰患者中应用的安全性和有效性。
1 研究对象和方法
1.1 研究对象 2008-2011年,在我院心内科住院的符合入选和排除标准,并确诊为射血分数保留(EF>45%)的心衰患者80例,随机分为2组,A组37例,B组43例。以上两组临床资料比较差异无统计学意义,P>0.05,见表1。
表1 两组入选病例基线资料情况()
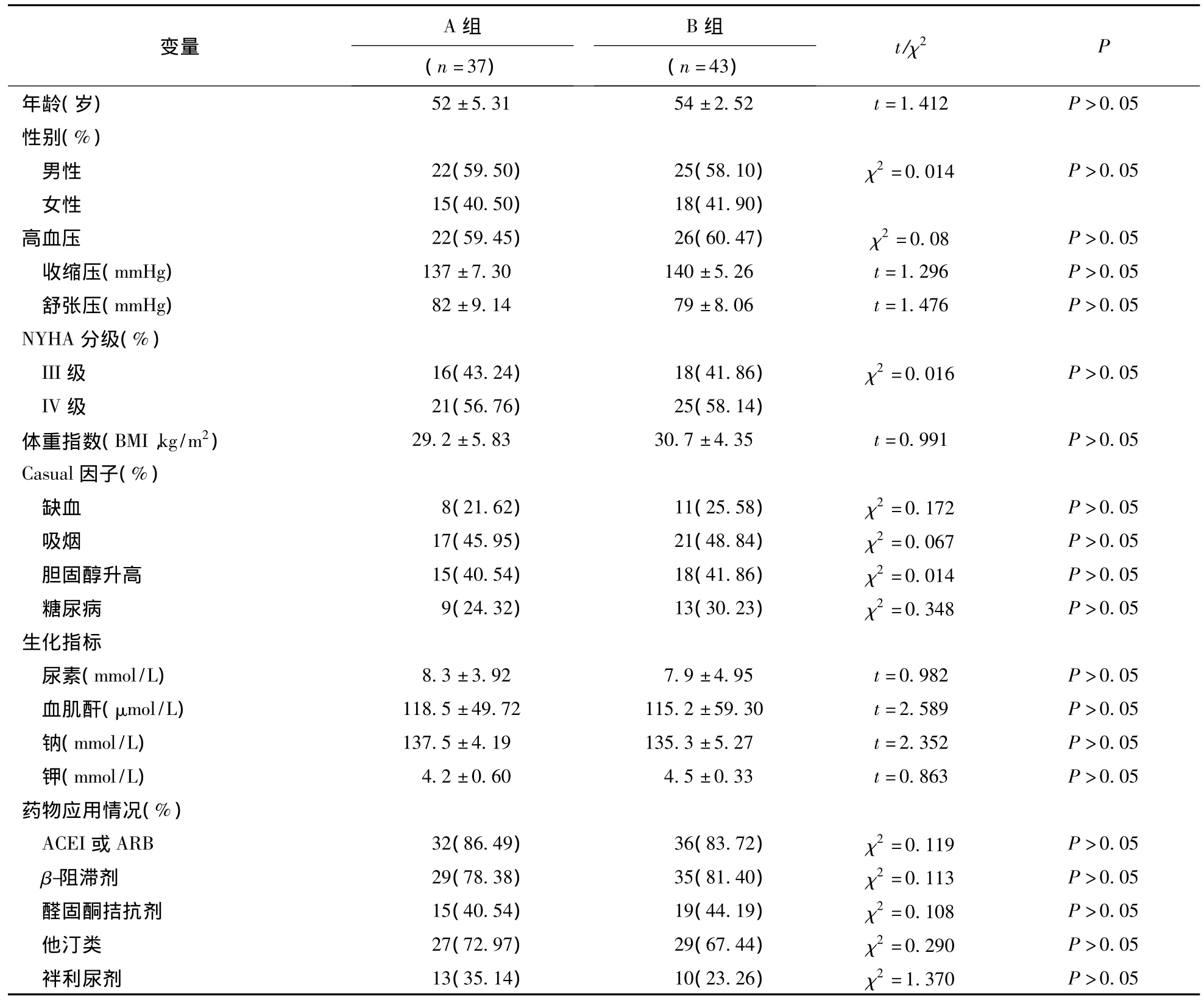
表1 两组入选病例基线资料情况()
注:BMI:体重指数-body mass index;ACEI:血管紧张素转换酶抑制剂;ARB:血管紧张素II受体阻滞剂
A 组B 组变量t/χ2P(n=37)(n=43)年龄(岁) 52±5.31 54±2.52 t=1.412 P >0.05性别(%)男性 22(59.50) 25(58.10) χ2=0.014 P>0.05女性 15(40.50) 18(41.90)高血压 22(59.45) 26(60.47) χ2=0.08 P>0.05收缩压(mmHg) 137±7.30 140±5.26 t=1.296 P>0.05舒张压(mmHg) 82±9.14 79±8.06 t=1.476 P>0.05 NYHA分级(%)III级 16(43.24) 18(41.86) χ2=0.016 P>0.05 IV级 21(56.76) 25(58.14)体重指数(BMI,kg/m2) 29.2±5.83 30.7±4.35 t=0.991 P >0.05 Casual因子(%)缺血 8(21.62) 11(25.58) χ2=0.172 P>0.05吸烟 17(45.95) 21(48.84) χ2=0.067 P>0.05胆固醇升高 15(40.54) 18(41.86) χ2=0.014 P>0.05糖尿病 9(24.32) 13(30.23) χ2=0.348 P>0.05生化指标尿素(mmol/L) 8.3±3.92 7.9±4.95 t=0.982 P>0.05血肌酐(μmol/L) 118.5±49.72 115.2±59.30 t=2.589 P>0.05钠(mmol/L) 137.5±4.19 135.3±5.27 t=2.352 P>0.05钾(mmol/L) 4.2±0.60 4.5±0.33 t=0.863 P>0.05药物应用情况(%)ACEI或 ARB 32(86.49) 36(83.72) χ2=0.119 P >0.05 β-阻滞剂 29(78.38) 35(81.40) χ2=0.113 P>0.05醛固酮拮抗剂 15(40.54) 19(44.19) χ2=0.108 P>0.05他汀类 27(72.97) 29(67.44) χ2=0.290 P>0.05袢利尿剂 13(35.14) 10(23.26) χ2=1.370 P >0.05
1.2 入选标准 左室射血分数>45%;胸部X片心胸比<0.5;纽约心功能分级III~IV(NYHA分级);年龄在45~65岁之间。
1.3 排除标准 心肌病:扩张型心肌病、肥厚型心肌病、限制型心肌病;瓣膜心脏疾病:主动脉缩窄、主动脉瓣狭窄反流;心包疾病:心包炎、心包积液;慢性阻塞性肺部疾病、肝硬化和肾功能衰竭等;患有其他严重疾病,不愿或不适合参与者。
2 方法
2.1 超声指标测量方法 选用美国GE公司Vivid7型高档彩色超声心动图仪,探头频率为4SMHz,计算左室射血分数,标准方法测量心脏心尖四腔,采用 Simpson法,测3次取平均值;-dp/dtmax的测量方法为:用二尖瓣反流的下降最大速率进行估测,在二尖瓣反流血流多普勒频谱的减速支中,每隔22 ms测量反流速度,用简化的伯努利方程换算成压差PG,然后测量减速支中每两点间的压差下降速率,然后取最大值。测出左室最大压力下降速率(-dp/dtmax),再测出该值下降时的两点间的压力差p0,把两个数据同时代入此公式T=-p0/(-dp/dtmax),计算出左室压力下降时间常数(T),而与(-dp/dtmax)相比,左室压力下降时间常数(T)更能客观反映左室舒张功能。
2.2 BNP的测定方法 所有两组患者取(禁食12 h,空腹抽血)静脉血2mL,加入未应用其他添加剂的依地酸二钠钾(EDTA)抗凝试管中,分别在入院用药前、治疗2周后,测量患者血清BNP浓度值。测量方法采用电化学发光法测定血清BNP浓度值(试剂盒及仪器均来自美国罗氏公司ELECSYS2010),严格依据试剂说明书来规范化操作。
2.3 治疗方法 A组给予纠正诱因,单纯传统抗心衰药物(利尿剂、硝酸酯类、β受体阻滞剂、ACEI/ARB类)对症治疗,具体剂量以纠正患者症状及诱因为标准;B组在A组药物治疗的基础上加用法舒地尔。法舒地尔给药方法:剂量为30 mg/次,2次/d,加入100mL生理盐水或5%葡萄糖250mL注射液,非同次持续静脉滴注30min以上,连续应用14 d,采用即用即配液,暂时不用时保存于-80°冰箱,以防失效。法舒地尔制剂均由同一厂家提供,选用同一批次或相邻批次制剂。
2.4 统计学处理 计数资料采用卡方检验,计量资料采用t检验;组间数值比较采用独立样本t检验,治疗前后采用配对样本t检验。由于超声心动图示左室压力最大下降速率(-dp/dtmax)及左室压力下降时间常数(T)等参数均不符合正态分布,故先用Levene方差齐性检验,然后进行t检验。均采用双侧检验,P<0.05为差异有统计学意义,P≤0.01为有极显著统计学意义。所有数据采用SPSS16.0统计软件包处理。
3 结果
3.1 入院2周后两组患者NYHA分级变化情况见表2。

表2 入院2周后两组患者NYHA分级变化情况
3.2 出院4周后两组患者NYHA分级变化情况 见表3。

表3 出院4周后两组患者NYHA分级变化情况

表4 出院6周后两组患者NYHA分级变化情况
3.3 两组治疗前,治疗2周后BNP值组间、组内比较情况 见表5。
3.4 两组治疗前,治疗2周后-dp/dtmax值组间、组内比较 见表6。

表5 两组治疗前、治疗2周后BNP值组间、组内比较情况

表6 两组治疗前,治疗2周后-dp/dtmax值组间、组内比较
3.5 超声指标左室压力下降时间常数(T)治疗前后及组间比较 见表7。

表7 超声指标左室压力下降时间常数(T)治疗前后及组间比较
4 讨论
HFPEF是由于心脏心室舒张功能受损、顺应性减低,心室壁僵硬度增高,导致心室舒张期血液充盈受损或下降(主要为左心室),心室舒张末压力增高,进而导致心输出量减低,加剧血液循环动脉系统的相对供血不足,最终出现心力衰竭。HFPEF的心脏组织结构特点为:心肌细胞适应不良性肥大、心室壁显著的增厚(尤以室间隔和左室后壁为著)、心室腔容积大小正常、射血分数保留(EF>45%)并伴有心室充盈受限。心室压力、容积超负荷可能会影响心脏舒张期的时间、速率以及心肌松弛的完成[5-6]。HFPEF主要特点是左心室几何形态和组织结构的改变,此类病理改变被称为心脏重构(Cardiac remodeling,CR)。心脏重构往往发生于心衰临床表现出现前(可持续数月至数年),在出现明显症状后病程仍将持续进展[7],虽然给予改善乃至最佳药物强化治疗,其病程及临床表现仍继续逐步恶化[8]。
一般认为,在HFPEF产生的病理生理机制中,心脏局部纤维化激活过程可能作为一种重要的机制参与其中,纤维化使得心室的僵硬度增加、舒张功能受损、进而充盈受限,最后导致心力衰竭的发生;心肌纤维化为高血压肥厚性心肌病的标志,同时也是心脏性猝死、室性快速型心律失常及左室舒张功能不全的重要基础[9];而心脏纤维化在心肌间质则更显著,正常心肌间质中的主要胶原纤维为I型及III型,胶原纤维首先在成纤维细胞内合成前体,然后释放入细胞外基质,分解为胶原纤维[10]。I型前胶原羧基末端肽(Procollagen type I carboxyterminal peptide,PICP)为I型胶原纤维分解的产物,生理情况下其产生量与释放入血由肝脏清除量的比值为1∶1,能可靠地反映I型胶原的合成[11-12];如果该比值增大提示胶原合成增加,其直接结果是心肌胶原纤维沉积增加,从而为心脏心室壁几何形态和力学特点的改变奠定病理学基础。而在舒张功能受损的早期,虽然无MRI等影像组织学信号的改变,但已有胶原纤维代谢异常激活,PICP等胶原代谢血清标志物升高。Lopez[13]等研究证实,HFPEF患者心肌总胶原容积显著升高。实验室[14]及临床[15-17]数据表明,心肌纤维化与心室舒张功能异常及心肌僵硬直接相关,并且针对降低心室纤维化的治疗可改善舒张功能;HFPEF患者心室的僵硬度的增加及顺应性的下降,势必导致心室壁的张力显著升高,进而引起心室壁释放BNP,同时引起血清及心肌局部神经内分泌因子的激活并升高,因此,此类因子在一定程度上可以反映心室壁舒张功能状态。BNP浓度的变化与心力衰竭的病情程度变化一致,两类心力衰竭均有此类改变,即BNP能随着心功能的改善而下降[18];心肌细胞外基质过度胶原组织形成为特点的病理改变,是心室舒张功能恶化并最终导致心衰的病因[19];心肌纤维化结果的产生,不仅与刺激胶原合成增加有关,而且同时与胶原纤维降解不变或被抑制有关。而高血压所致的心肌纤维化,同样可能为基质金属蛋白酶对这些胶原分子清除分解不足所致。基质金属蛋白酶(MMPs)广泛参与了细胞外基质的降解[20],其在心肌细胞外基质尤其是胶原纤维的降解中起重要的作用,其基因表达的增加及其活性的增强,对心肌细胞外基质胶原纤维的堆积有着显著的作用[21-22];与此同时,Rho激酶在心肌细胞及间质成纤维细胞的多种功能中同样起着重要的作用,如肌动蛋白细胞骨架的构成和细胞的粘附迁移[19]。有研究显示,小分子GTP酶Rho家族和MMPs家族成员对肌动蛋白应力纤维及黏着斑的快速聚集均有调节作用[23],而后两者是细胞外基质降解和细胞迁移过程所必备的先决条件[24-25]。佟浩等研究发现[26],随着心肌重塑加重、心功能的受损加重,RhoA、Rho激酶及MMP-3、MMP-9基因表达显著增加,且前两者与后两者的基因表达呈正相关趋势,并认为RhoA通过Rho激酶结合,刺激MMP-3、MMP-9基因表达引起细胞外基质的结构发生改变,进而引起心肌重塑和心功能恶化;Rho激酶抑制剂-法舒地尔能抑制Rho信号分子诱导的通路,通过抑制应力纤维的合成,进而抑制心肌组织局部纤维化。有研究显示,在成年大鼠心肌中,压力超负荷可诱导Rho激酶通路的迅速激活,以此推测:有机械应力触发的心肌细胞最初适应性改变及其机制的协调中,Rho激酶通路可能起关键作用[27]。
基于以上基础理论的研究,本研究分别通过纵向比较A、B两组入院用药前、用药2周后患者的血清BNP值,以及左室压力下降时间常数(T)、左室压力下降最大速率(-dp/dtmax)等指标,发现A、B两组在药物干预2周后,在临床症状改善的基础上,以上指标均有显著的改善;而法舒地尔组对血清BNP的干预作用更强,法舒地尔组患者的浓度降低更显著。相关指标改善表现为,血清BNP值较入院患病时下降,左室压力下降时间常数(T)缩短;同时,横向比较,即传统药物治疗组与法舒地尔干预组相比,左室舒张功能超声指标左室压力下降时间常数(T)可以进一步缩短,两组间有统计学意义(P<0.05)。虽然加用法舒地尔组与传统药物治疗组对两组患者的左室压力下降最大速率(-dp/dtmax)值均有改善作用,但两组之间尚未发现有明显统计学差异。分析认为:由于临床中实际测量的(-dp/dtmax)值为发生在主动脉瓣关闭稍后时间的左心室压力下降最大速率,对其干扰的因素较多,诸如受左室收缩压(LVSP)、左室收缩末期容积(LVESV)、主动脉瓣关闭不全(AR)、主动脉压(AP)、回心血量、心率(HR)以及Ca2+在心肌细胞摄取、结合等因素的影响,因此,解读该指标时应当更加审慎,只能作为部分参考指标;血清BNP作为一项心力衰竭的实验室指标,受体内多种体液因子及机体不同功能状态调节,此外实验室检测的误差同样应考虑在内。倘若射血分数保留的心衰中老年患者,传统药物最优化治疗不能得到进一步改善的情况下,适当应用Rho激酶抑制剂,不失为一种较好的选择。
[1]Burridge K,Wennerberg K.Rho and Rac take center stage[J].Cell,2004,116(2):167-169.
[2]Takeuchi S,Kawashima S,Rikitake Y,et al.Cerivastatin suppresses lipopolysaccharide-induced ICAM-1expression through inhibition of Rho-GTPase in BAEC[J].Biochem Biophy Res Commun,2000,269(1):97-102.
[3]赵慧颖,马小欣,陈冬梅,等.法舒地尔对大鼠动脉粥样硬化斑块中单核细胞趋化蛋白-1表达的抑制作用[J].Chin Microcirc,2009,13:344-347.
[4]汪祥海.Rho激酶在血管紧张素Ⅱ刺激大鼠心肌成纤维细胞增殖和胶原合成中的作用[J].中国病理生理杂志,2007,23(6):1098-1101.
[5]De Hert SG,Gillebert TC,Ten Brorcke PW,et al.Length-dependent regulation of left ventricular function in coronary surgery patients[J].Anesthesiology,1999,91:379-387.
[6]Leite-Moreira AF,Correia-Pinto J,Gillebert TC,et al.Afterload induced changes in myocardial relaxation:A mechanism for diastolic dysfunction[J].Cardiovasc Res,1999,43:344-353.
[7]徐全波,樊永平,刘永欣,等.急性心肌梗死后左心室重构的研究进展[J].中国医药,2008,3(2):124-126.
[8]Jessup Mariell,Abraham William T,Chin Marshall H,et al.2009 Focused Update Incorporated into the ACCF/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults[J].JACC,2009,53(15):e7-9.
[9]Ho Carolyn,Lopez Begona,Otavio R,et al.Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy[J].NEJM,2010,363:552-563.
[10]黄斌雄.急性心肌梗死患者血浆II型前胶原氨基末端肽含量的变化[J].临床心血管病杂志,2002,18(4):181-182.
[11]Querejeta R,López B,González A,et al.Increased collagen type I synthesis in patients with heart failure of hypertensive origin:relation to myocardial fibrosis[J].Circulation,2004,110:1263-1268.
[12]Martos Ramom,Baugh John,Ledwidge Mark,et al.Evidence of increased myocardial collagen turnover linked to diastolic dysfunction[J].Circulation,2007,115:888-895.
[13]López B,González A,Querejeta R,et al.Alteration in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure[J].Am Coll Cardiol,2006,48:89-96.
[14]Diez J,Panizo A,Gil MJ,et al.Serum Markers of collagen type I metabolism in spontaneously hypertensive rats:relation to myocardial fibrosis[J].Circulation,1996,93:1026-1032.
[15]Querejeta R,Lopez B,González A,et al.Increased Collagen Type I Synthesis in Patients with heart failure of Hypertensive Origin Relation to Myocardial Fibrosis[J].Circulation,2004,110:1263-1268.
[16]Brilla CG,Funk RC,Rupp H,et al.Lisinopril-Mediated regression of myocardial fibrosis in Patients with Hypertensive heart disease[J].Circulation,2000,102:1388-1393.
[17]Diez J,Querejeta R,Lopez B,et al.Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients[J].Circulation,2002,105:2512-2517.
[18]Cheng V,Kazanagra R,Garcia A,et al.A rapid bedside test for B-type peptide predicts treatment outcomes in patients admitted for decompensated heart failure:a pilot study[J].Am Coll Cardiol,2001,37(2):386-391.
[19]Shimokawa H.Rho-kinase as a novel therapeutic target in treatment of cardiovascular disease[J].Cardiovasc Pharmacol,2002,39:319-327.
[20]吉洁,沈光宇.基质金属蛋白酶9在脑出血中作用的研究进展[J].实用医学杂志,2009,25(24):4244-4245.
[21]田飞,阿丽亚,张新平.基质金属蛋白酶与动脉粥样硬化关系的研究进展[J].中国医药,2010,5(11):1101-1102.
[22]戴贵军,孙建辉,白江涛,等.血清S100A8/A9、MMP-2水平变化与急性冠状动脉综合征的关系[J].实用医学杂志,2010,26(24):4528-4530.
[23]Sounni NE,Noel A.Membrane type-matrix metalloproteinases and tumor progression[J].Biochimie,2005,87:329-342.
[24]Jaffe AB,Hall A.Rho-GTPases:Biochemistry and biology[J].Annu Rev Cell Dev Biol,2005,21:247-269.
[25]Vicente-Manzanares M,Webb DJ,Horwitz AR,et al.Cell migration at a glance[J].Cell Sci,2005,118:4917-4919.
[26]佟浩,张曼.RhoA经基质金属蛋白酶-3、-9通路介导心力衰竭大鼠心肌重塑及心功能恶化[J].解剖学杂志,2008,31(2):173-176.
[27]Torsoni AS,Fonseca PM,Crosara-Alberto DP,et al.Early activation of p160ROCK by pressure overload in rat heart[J].Am J Physiol Cell Physiol,2003,284:C1411-C1419.
