微生物谷氨酰胺转胺酶的表达及分子改造研究进展
2011-02-09刘松张东旭堵国成陈坚
刘松,张东旭,堵国成,陈坚
江南大学生物工程学院 工业生物技术教育部重点实验室,无锡 214122
微生物谷氨酰胺转胺酶的表达及分子改造研究进展
刘松,张东旭,堵国成,陈坚
江南大学生物工程学院 工业生物技术教育部重点实验室,无锡 214122
微生物谷氨酰胺转胺酶具有催化蛋白质和某些非蛋白物质交联的功能,被广泛应用于食品、医药及纺织等领域。为提高该酶的产量及建立相应的分子改造平台,上世纪90年代日本味之素公司便开展了微生物谷氨酰胺转胺酶重组菌构建的研究。目前,该酶已在多个表达系统中实现活性表达,部分重组菌较野生菌的产酶能力有显著提高。近年来,谷氨酰胺转胺酶的分子改造研究也取得了初步进展,酶的催化活力、热稳定性及底物专一性得到提升。文中对上述研究中涉及的蛋白质表达及改造策略进行了简要的总结及分析,并指出相关研究的发展趋势。
链霉菌,谷氨酰胺转胺酶,蛋白质表达,分子改造,蛋白质结构
TGase最初从动物[3]和植物组织[4]中提取,产量低,分离成本高。1989年,日本味之素公司首次在土壤中分离到 TGase生产菌株,茂原链轮丝菌Streptoverticillium mobaraense S-8112[5]。此后,研究者不断分离到新的TGase生产株菌,包括拉达克轮丝菌 Streptoverticillium ladakanum[6],吸水链霉菌Streptomyces hygroscopicus[7],环状芽胞杆菌Bacillus circulans[8],枯草芽胞杆菌Bacillus subtilis[9]等。与其他来源的TGase相比,微生物发酵生产的TGase具有非Ca2+依赖性的特点,且生产周期短,成本优势明显。目前,链霉菌TGase已广泛用于食品工业,适量添加TGase可显著改善各类食品蛋白质的功能性质和营养价值,应用范围包括肉制品、水产品、乳制品、植物蛋白制品等[1]。此外,TGase在一些非食品工业领域也具有较大的应用前景,如组织工程、纺织工业及生物学研究等[2]。然而,依靠优势菌种选育和过程优化[10-11]的传统发酵技术对TGase产量的提高十分有限,远不能满足市场需求。
为进一步提高产量,日本味之素公司率先开展了 TGase重组菌构建的工作。随后,德国Martin-Luther University、台湾食品工业发展研究所、台湾国立中兴大学、中科院微生物研究所、华南理工大学及诺和诺德 (中国) 制药有限公司等相关研究机构或企业相继报道了重组TGase的制备策略。至此,链霉菌TGase已在大肠杆菌、酵母、链霉菌及谷氨酸棒杆菌等宿主中成功表达(表1)。其中,重组谷氨酸棒杆菌胞外TGase活力达到16.3 U/mL[12],远高于野生菌发酵产酶水平[10]。然而,特有的催化性能[1]及活化机制[13]使链霉菌TGase在异源宿主中的高效表达遇到诸多问题,如,低表达量、易形成包涵体及无催化活性等。本文对上述研究中涉及的蛋白质表达策略进行了简要的分析,并总结了TGase分子改造研究的最新进展,旨在为微生物 TGase的后续研究及具有类似活化机制的功能蛋白或酶类[14]的表达与改造提供参考。
1 重组TGase表达
如图2所示,链霉菌TGase是一种胞外酶,以无活性的酶原 (pro-TGase) 形式分泌,pro-TGase被切除N-端酶原区后,转化成活性TGase[15]。在分泌过程中,金属蛋白酶 (TAMEP) 及丝氨酸蛋白酶参与了TGase的活化[13,16]。链霉菌TGase基因的完整读码框由信号肽基因、酶原区基因及成熟酶基因组成[6,15,17-19]。基于此,TGase表达系统的构建围绕TGase及pro-TGase的表达进行,历经了TGase非活表达及活性表达两个阶段。
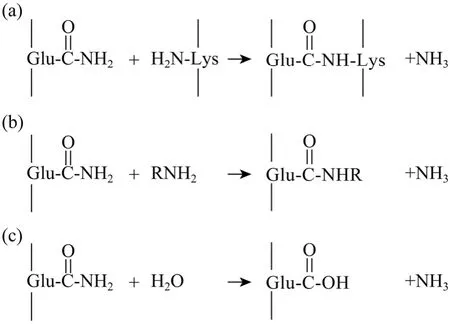
图1 TGase催化反应[1]Fig. 1 The catalytic reactions mediated by TGase[1].
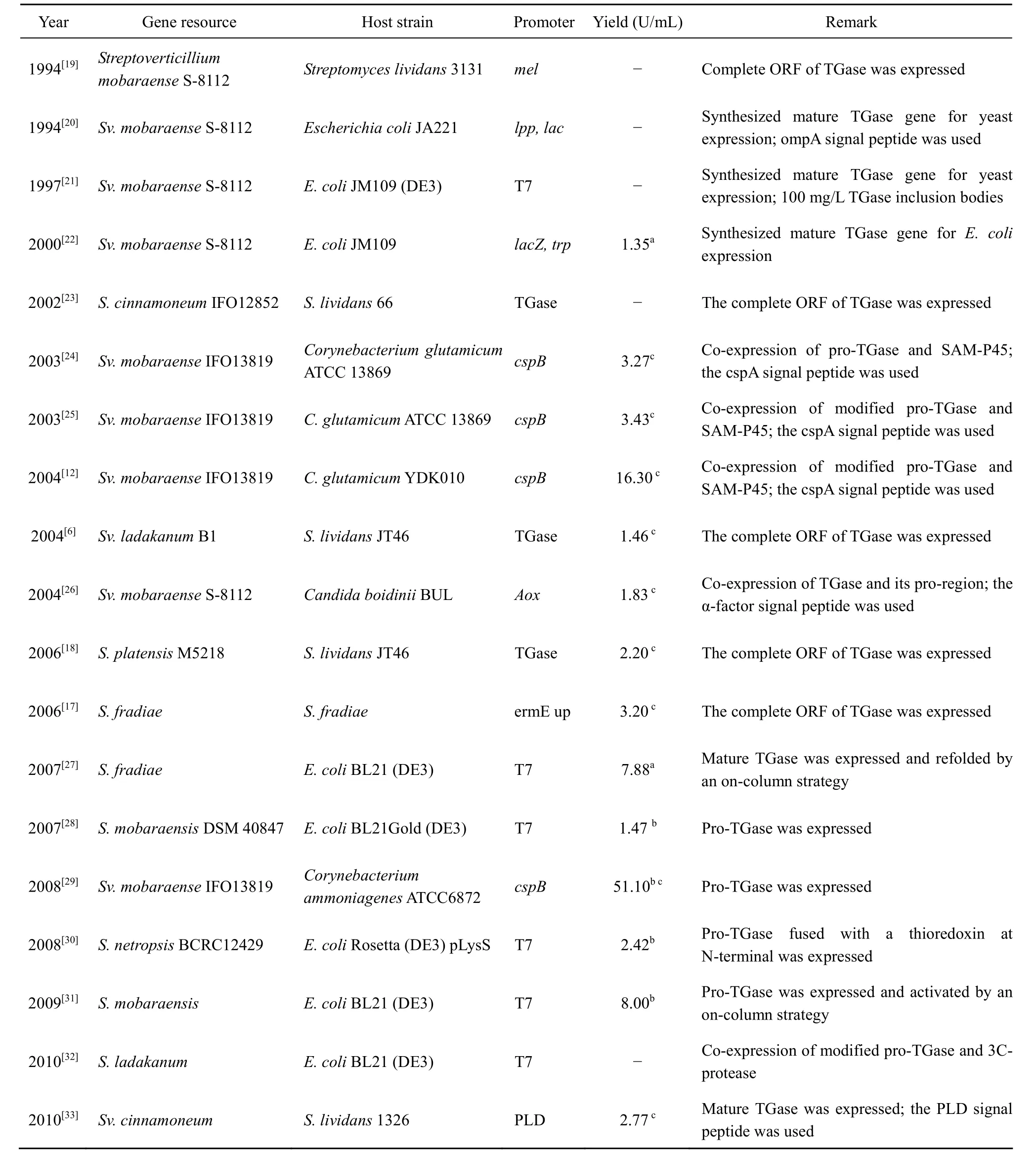
表1 链霉菌TGase在异源体系中的表达Table 1 Expression of Streptomyces TGase in heterogeneous system
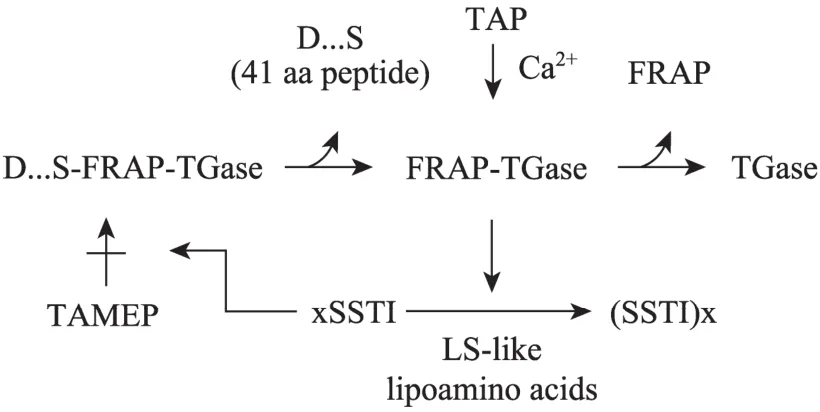
图2 链霉菌pro-TGase活化机制[13]Fig. 2 The mechanism of pro-TGase activation[13].
1.1 TGase的非活性表达
1.1.1 表达TGase成熟酶
表达TGase成熟酶基因是获得重组TGase最直接的方法。在大肠杆菌中,N-端融合信号肽的TGase可分泌至周质空间,但是蛋白表达量极低,且比酶活力远低于野生酶[20]。而换用强启动子[21]、N-端融合表达增强序列[21]以及密码子偏好性优化[22]虽可提高表达量,但TGase基本以包涵体形式存在。使用受严谨调控的T7启动子仍无法解决TGase成熟酶在大肠杆菌中的可溶性问题[27]。令人意外的是,虽然TGase成熟酶在酵母中成功分泌至胞外,却无法获得催化活性[26]。因此,上述重组TGase需经过体外复性才能获得催化活性。传统的稀释法复性得到的TGase比酶活力可达到野生酶的水平,总酶活力收率达到50%[34]。“双梯度 (脲浓度与pH) 上柱复性”同样可成功复性 TGase[27]。然而,蛋白复性的高成本不利于TGase工业规模的生产。
1.1.2 表达pro-TGase
与TGase成熟酶的表达不同,各链霉菌来源的pro-TGase均可在大肠杆菌中高效可溶表达[28,30]。采用葡萄糖流加策略进行重组菌的高密度发酵,pro-TGase产量显著提高[31]。结果表明,TGase酶原区的存在可能提高了 TGase在大肠杆菌中的溶解性。同样,pro-TGase亦可在链霉菌、酵母及棒杆菌中高效可溶表达。在变铅青链霉菌 Streptomyces lividans 66中,pro-TGase在自身启动子及信号肽介导下即可高效分泌[23]。以酵母或棒杆菌为宿主时,pro-TGase则需在N-端融合宿主信号肽才能被分泌至胞外[26,29]。强化pro-TGase分泌的策略有2种:1) 修饰 pro-TGase酶原区;2) 优化信号肽与宿主菌的组合。前者可使pro-TGase分泌量提高25%[12],后者得到的胞外pro-TGase可达到2 500 mg/L,是目前报道的最高水平[29]。
Pro-TGase表达后需经切除酶原区后才能转化成活性TGase。用于pro-TGase活化的蛋白酶可从产TGase的野生菌 (如 Sv. mobaraense分泌的TAMEP[35]) 或其他链霉菌 (如白浅灰链霉菌Streptomyces albogriseolus分泌的 SAM-P20、SAM-P26及 SAM-P45等[23]) 中提取。一些商品化的蛋白酶亦能活化pro-TGase,如中性蛋白酶及胰蛋白酶等[36]。然而,活化反应体系中残存的蛋白酶活力可能持续降解活性TGase,不利于其保存和应用[36]。为消除蛋白酶存留的不利影响,采用“上柱激活”的方法可将残余的活化蛋白酶与TGase分离,TGase贮存稳定性显著提高[31]。
1.2 TGase的活性表达
1.2.1 选择适宜的宿主
在多数表达宿主中,TGase需要其酶原的辅助才能高效可溶表达。然而,特定的宿主环境似乎可以消除 TGase对酶原区的这种依赖。最新的报道表明,在肉桂链轮丝菌 Streptoverticillium cinnamoneum磷脂酶D (PLD) 启动子及其信号肽的介导下 Sv. cinnamoneum TGase成熟酶基因可在 S. lividans 1326中高效表达 (230 mg/L),产物分泌至胞外且比酶活力与野生酶相当[33]。
选用分泌活化蛋白酶的宿主表达 pro-TGase是直接生产活TGase的另一种方式。已发现具有TGase活化蛋白酶的宿主主要是链霉菌,包括 S. lividans3113[19]及 S. lividans JT46[6]。其中,以 S. lividans JT46为表达宿主时,TGase的活性表达较为成功。Sv. ladakanum[6]及平板链霉菌 Streptomyces platensis[18]来源的 pro-TGase在其内源启动子及信号肽的作用下均可分泌至胞外;前者只有部分转化为活性 TGase (1.46 U/mL) 而后者则完全活化(2.2 U/mL)。活化效率的差异可能是由于pro-TGase氨基酸序列不同导致的,后者的酶原区切割位点可能更易于被宿主蛋白酶所识别。因此,在酶原区与TGase成熟之间引入宿主蛋白酶特异性切割位点有望提高活性TGase的产量。事实上,在酶原区C-端加入酵母 Kex2肽链内切酶识别位点后,pro-TGase在酵母中表达后可直接转化成活性 TGase[26]。值得注意的是,将 pro-TGase克隆至原始菌株中表达,借助其野生活化系统进行活化是一种提高TGase产量的有效方式[17]。
1.2.2 共表达活化蛋白酶
与其他表达策略相比,在无活化蛋白酶的宿主中共表达 pro-TGase及其活化蛋白酶得到活性TGase产量最高。在谷氨酸棒杆菌中共表达分别融合分泌信号肽的SAM-P45及pro-TGase,大量活性TGase即可分泌至胞外[24];改造酶原区C-端后,该体系即可得到带有天然N-端的TGase[25]。此外,共表达人鼻病毒3C蛋白酶及加入3C蛋白酶识别位点的 pro-TGase同样可在大肠杆菌中直接得到活性TGase[32]。然而,共表达体系产生的活化蛋白酶存在持续降解活性TGase的可能[24],同时蛋白酶活力对TGase产品的应用也将造成不利影响。
1.2.3 共表达酶原区
如前所述,在表达过程中酶原区对TGase的可溶性及催化活性有辅助作用。这种辅助作用可通过分子内相互作用来完成,即表达pro-TGase;也可通过分子间相互作用实现,即共表达酶原区与 TGase成熟酶。共表达酶原区与TGase成熟酶的一个重要优势是无需活化蛋白酶参与即可得到活性 TGase,可避免共表达活化蛋白酶的不利影响。到目前为止,该策略只在酵母系统中成功实施[26]。
2 TGase分子改造
基因水平上的分子改造需要易于操作的蛋白表达系统及准确可行的检测方法。应用于TGase分子改造的表达系统主要是大肠杆菌[28,32]及谷氨酸棒杆菌系统[37]。适于突变体筛选的TGase活力检测方法包括三肽为底物的显色法[37]、可定量检测大分子交联反应的基于酶标技术 (ELT) 的核磁共振法[38]及邻近闪烁分析法 (SPA) 等[32]。此外,应用反向HPLC检测蛋白浓度的技术为TGase比酶活力分析提供了快速可靠的方法[37]。目前,分子改造主要以茂原链霉菌Streptomyces mobaraensis (即Sv. mobaraense)来源的TGase为研究对象。
2.1 理性设计
2.1.1 基于分子柔性区域分析的定点突变
异核单量子相干谱 (HSQC) 可检测酶分子中特定区域氨基酸缺失或突变对催化活性中心构象的影响;若缺失或突变未导致酶分子错误折叠但改变了催化活性中心的构象,则表明该区域具有一定的柔韧性,可作为分子改造的目标区域[39]。研究发现,N-端缺失第一个氨基酸 (Asp) 的 TGase (Ser-TGase) 较野生酶具有更高的催化活力[38]。基于此,通过HSQC确定N-端是TGase的一个柔性区域。实验表明,适度缺失及取代突变N-端氨基酸残基均可提高TGase的催化活性[39]。该方法可对晶体结构未知的酶分子进行理性改造,但柔性区域的检测仍具有偶然性,需要一定的前期研究基础。
2.1.2 基于水接触表面热点区域的定点突变(WASH-ROM)
WASH-ROM 认为,酶分子中具有高溶剂触表面积 (SAS) 且与催化活性中心临近的氨基酸残基可能影响酶与底物的接触,通过突变这些氨基酸可强化酶与底物的接触,从而提高其催化活性[37]。在TGase分子的改造过程中,利用生物信息学软件,如Discovery Studio (Accelrys Software Inc.),计算出离TGase催化活性中心15Å以内的90个氨基酸残基的相对SAS,选取其中40个具有高相对SAS的氨基酸残基分别进行1~6种氨基酸的单点取代[37],得到的突变体最高比酶活力达到41.7 U/mg,是野生TGase比活力的1.5倍。该改造策略较前者更高效,但必须已知酶分子的晶体结构。
2.2 定向进化
理性设计改造TGase的突变体筛选工作量少,但其理论依据仍然有限。随机突变的筛选工作量大,但可以对TGase多方面的催化性能进行改造,如热稳定性、热敏性及催化活力等。定向进化改造TGase的关键是建立突变库高通量筛选方法。以大肠杆菌表达体系为筛选平台时,细胞破碎、TGase的活化及酶活力检测均在多孔板上完成[40]。通过随机突变,正突变体的热稳定性达到野生TGase的2.7倍,而热敏性突变体则只保留了10 ℃~40 ℃间的催化活性[40]。以谷氨酸棒杆菌为筛选平台时,TGase突变体基本分泌至胞外,无需细胞破碎及 TGase活化即可在多孔板上完成活性的筛选,工作效率显著提高;所得 TGase正突变体最高比活力达到42.9 U/mg,是野生酶的1.7倍[37]。
2.3 理性设计与定向进化的结合
理性设计与随机突变结合的基本思想是,根据已知数据和知识选择酶分子特定区域进行随机突变,在不影响突变效果的前提下降低突变库的规模,可显著提高分子改造的效率。改造TGase底物专一性的同时,发现N-端带有额外Gly及Pro的TGase (Gly-Pro-TGase) 对人类生长因子 Gln141催化作用较野生TGase具有更高的专一性[32];晶体结构分析表明Tyr62及Tyr75可能影响TGase的催化活力,且两者的取代突变 (Tyr62His及 Tyr75Phe) 可提高TGase对 Gln141的专一性。基于此,对 Gly-Pro-TGase中Tyr62及Tyr75分别进行17种及12种氨基酸取代,构建饱和突变库,最终筛选得到5个TGase突变体对Gln141具有高度专一性[32]。
2.4 TGase的结构与功能
晶体结构显示 (图3),S. mobaraense TGase的二级结构属于α+β型,含11个α-螺旋及8个β-折叠;分子总体呈盘状并带有一个裂缝,两个loop环(Asp1-Ala10与Asn276-Met288) 组成裂缝左壁,一个loop环 (Asn239至Asn253) 组成裂缝右壁;催化活性基团Cys64、Asp255及His274均位于分子裂缝底部[41]。随着分子改造研究的推进,TGase分子中影响酶学性质的重要结构区域得到初步揭示,包括:
1) 分子裂缝的左壁区域。该区域中N-端 loop环的修饰对 TGase催化性质有较大影响。适度缺失 N-端氨基酸残基 (1~3个) 可提高 TGase的催化效率[39]。这可能是由于氨基酸的缺失降低了底物与催化活性中心接触的空间位阻。N-端紧靠裂缝的氨基酸取代 (Ser2Arg等)[39]及位于分子表面的极性氨基酸取代 (Ala10Ser等)[37]或酸性氨基酸取代(如,Glu28Asp等)[37]均可提高TGase的催化活性。此外,N-端的单点突变还可提高 (Ser2Phe等) 或降低 (Pro9Leu等) TGase的热稳定性[40]。
2) 分子表面与裂缝临近区域。该区域包括盘状分子的正面及背面。分子正面 (Gln74Ala等) 及背面 (Ser299Phe等) 的疏水性氨基酸取代突变提高了TGase催化活性[37]。而位于分子正面与裂缝临近的Tyr62及Tyr75的取代突变还可显著提高TGase的底物专一性[32]。
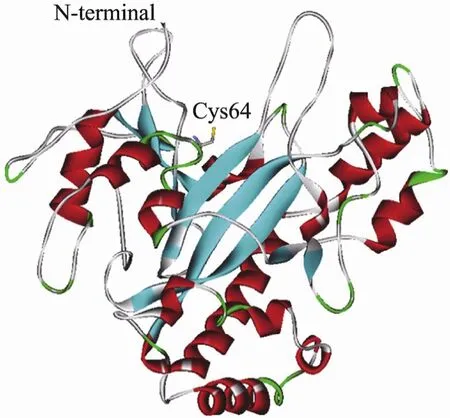
图3 S. mobaraense TGase晶体结构 (正面)[41]Fig. 3 Crystal structure of S. mobaraense TGase (front view)[41].
3) 分子内部与催化活性基团临近的区域。与催化活性基团 Asp25临近的Gly257Ser与 Lys269Glu可增强TGase的热稳定性[40]。分子内部的疏水性氨基酸取代 (Tyr42Phe及Ser199Ala) 对催化活性也有较大影响[37]。值得注意的是,N-端若添加Phe-Arg-Ala-Pro使 Ser199Ala的催化活性进一步提高[37],体现了在酶活力调节过程中该区域与N-端区域的协同效应。突变体的晶体结构分析将有助于解释这种效应。
最近,分子对接实验 (Molecular docking) 已模拟出TGase与底物的结合方式[42]。合成小分子底物CBZ-Gln-Gly在TGase分子裂缝中被拉伸,活性位点通过疏水相互作用与底物结合;当TGase与大分子底物 (酪蛋白等) 结合时,TGase的底物识别位点将在分子裂缝表面进一步扩展,体现了微生物TGase对底物的适应性。总之,对TGase结构与功能的深入了解将有助于探测分子改造的靶点,提高改造效率。
3 总结与展望
大量的研究表明,链霉菌TGase在多数异源宿主中高效可溶表达需要其酶原区的辅助,这种辅助作用的本质是对TGase折叠进行不同性质的抑制。在大肠杆菌系统中,酶原区抑制TGase无序折叠产生包涵体;在酵母表达体系中,抑制TGase错误折叠产生无活性蛋白;在棒杆菌中,酶原区则抑制TGase快速折叠从而促进其分泌。最近 pro-TGase的晶体结构[43]已经公布,为进一步分析酶原区的功能提供了结构基础。在已有的表达策略中,共表达TGase成熟酶及其酶原区较其他表达策略具有明显优势,是实现重组TGase工业规模生产的重要途径。在某些宿主中TGase无需酶原区亦可高效分泌,因此,筛选可直接分泌表达TGase成熟基因的宿主及信号肽同样应受到重视。
目前,微生物TGase分子改造的研究大部分以提高TGase的催化效率及热稳定性为目标,对其底物专一性的改造则关注较少。事实上,微生物TGase对底物具有较高的专一性。底物筛选实验表明,底物中谷氨酰胺残基的临近氨基酸组成[44-45]以及伯胺的链长[46-47]均对微生物 TGase催化反应有重要影响。随着微生物TGase应用领域的扩展,底物分子的多样性与TGase底物专一性的矛盾将日渐突出。因此,将来一个时期,在提高TGase环境稳定性的同时,应更加注重对TGase底物专一性的改造。近年来,理性设计与定向进化相结合的分子改造技术不断发展,如重复饱和突变 (ISM) 及组合活性位点饱和突变 (CASTing) 等[48],将为TGase的改造提供有力工具。
REFERENCES
[1] Yokoyama KI, Nio N, Kikuchi Y. Properties and applications of microbial transglutaminase. Appl Microbiol Biotechnol, 2004, 64(4): 447−454.
[2] Zhu Y, Tramper J. Novel applications for microbial transglutaminase beyond food processing. Trends Biotechnol, 2008, 26(10): 559−565.
[3] Aeschlimann D, Paulsson M. Transglutaminases: protein cross-linking enzymes in tissues and body fluids. Thromb Haemost, 1994, 71(4): 402−415.
[4] Serafini-Fracassini D, Del Duca S, Beninati S. Plant transglutaminases. Phytochemistry, 1995, 40(2): 355−365.
[5] Ando H, Adachi M, Umeda K, et al. Purification and characteristics of a novel transglutaminase derived from microorganisms. Agri Biol Chem, 1989, 53(10): 2613−2617.
[6] Lin YS, Chao ML, Liu CH, et al. Cloning and expression of the transglutaminase gene from Streptoverticillium ladakanum in Streptomyces lividans. Process Biochem, 2004, 39(5): 591−598.
[7] Cui L, Du GC, Zhang DX, et al. Thermal stability and conformational changes of transglutaminase from a newly isolated Streptomyces hygroscopicus. Bioresour Technol, 2008, 99(9): 3794−3800.
[8] De Barros Soares LH, Assmann F, Záchia Ayub MA. Purification and properties of a transglutaminase produced by a Bacillus circulans strain isolated from the amazon environment. Biotechnol Appl Biochem, 2003, 37(Pt 3): 295−299.
[9] Suzuki S, Izawa Y, Kobayashi K, et al. Purification and characterization of novel transglutaminase from Bacillus subtilis spores. Biosci Biotechnol Biochem, 2000, 64(11): 2344−2351.
[10] Zheng MY, Du GC, Chen J, et al. Modelling of temperature effects on batch microbial transglutaminase fermentation with Streptoverticillium mobaraense. World J Microbiol Biotechnol, 2002, 18(8): 767−771.
[11] Zheng MY, Du GC, Chen J. pH control strategy of batch microbial transglutaminase production with Streptoverticillium mobaraense. Enzyme Microb Technol, 2002, 31(4): 477−481.
[12] Date M, Yokoyama KI, Umezawa Y, et al. High level expression of Streptomyces mobaraensis transglutaminase in Corynebacterium glutamicum using a chimeric proregion from Streptomyces cinnamoneus transglutaminase. J Biotechnol, 2004, 110(3): 219−226.
[13] Schmidt S, Adolf F, Fuchsbauer HL. The transglutaminase activating metalloprotease inhibitor from Streptomyces mobaraensis is a glutamine and lysine donor substrate of the intrinsic transglutaminase. FEBS Lett, 2008, 582(20): 3132−3138.
[14] Eder J, Fersht AR. Pro-sequence-assisted protein folding. Mol Microbiol, 1995, 16(4): 609−614.
[15] Pasternack R, Dorsch S, Otterbach JT, et al. Bacterial pro-transglutaminase from Streptoverticillium mobaraense purification, characterisation and sequence of the zymogen. Eur J Biochem, 1998, 257(3): 570−576.
[16] Zhang DX, Wang M, Wu J, et al. Two different proteases from Streptomyces hygroscopicus are involved in transglutaminase activation. J Agri Food Chem, 2008, 56(21): 10261−10264.
[17] Liu XQ, Yang XQ, Xie FH, et al. Cloning of transglutaminase gene from Streptomyces fradiae and its enhanced expression in the original strain. Biotechnol Lett, 2006, 28(17): 1319−1325.
[18] Lin YS, Chao ML, Liu CH, et al. Cloning of the gene coding for transglutaminase from Streptomyces platensis and its expression in Streptomyces lividans. Process Biochem, 2006, 41(3): 519−524.
[19] Washizu K, Ando K, Koikeda S, et al. Molecular cloning of the gene for microbial transglutaminase from Streptoverticillium and its expression in Streptomyces lividans. Biosci Biotechnol Biochem, 1994, 58(1): 82−87.
[20] Takehana S, Washizu K, Ando K, et al. Chemical synthesis of the gene for microbial transglutaminase from Streptoverticillium and its expression in Escherichia coli. Biosci Biotechnol Biochem, 1994, 58(1): 88−92.
[21] Kawai M, Takehana S, Takagi H. High-level expression of the chemically synthesized gene for microbial transglutaminase from Streptoverticillium in Escherichia coli. Biosci Biotechnol Biochem, 1997, 61(5): 830−835.
[22] Yokoyama KI, Nakamura N, Seguro K, et al. Overproduction of microbial transglutaminase in Escherichia coli, in vitro refolding, and characterization of the refolded form. Biosci Biotechnol Biochem, 2000, 64(6): 1263−1270.
[23] Taguchi S, Arakawa K, Yokoyama KI, et al. Overexpression and purification of microbial protransglutaminase from Streptomyces cinnamoneum and in vitro processing by Streptomyces albogriseolus proteases. J Biosci Bioeng, 2002, 94(5): 478−481.
[24] Kikuchi Y, Date M, Yokoyama KI, et al. Secretion of active-form Streptoverticillium mobaraense transglutaminase by Corynebacterium glutamicum: processing of the pro-transglutaminase by a cosecreted subtilisin-like protease from Streptomyces albogriseolus. Appl Environ Microbiol, 2003, 69(1): 358−366.
[25] Date M, Yokoyama KI, Umezawa Y, et al. Production of native-type Streptoverticillium mobaraense transglutaminase in Corynebacterium glutamicum. Appl Environ Microbiol, 2003, 69(5): 3011−3014.
[26] Yurimoto H, Yamane M, Kikuchi Y, et al. The pro-peptide of Streptomyces mobaraensis transglutaminase functions in cis and in trans to mediate efficient secretion of active enzyme from methylotrophic yeasts. Biosci Biotechnol Biochem, 2004, 68(10): 2058−2069.
[27] Liu XQ, Yang XQ, Xie FH, et al. On-column refolding and purification of transglutaminase from Streptomyces fradiae expressed as inclusion bodies in Escherichia coli. Protein Expr Purif, 2007, 51(2): 179−186.
[28] Marx CK, Hertel TC, Pietzsch M. Soluble expression of a pro-transglutaminase from Streptomyces mobaraensis in Escherichia coli. Enzyme Microb Technol, 2007, 40(6): 1543−1550.
[29] Itaya H, Kikuchi Y. Secretion of Streptomyces mobaraensis pro-transglutaminase by Coryneform bacteria. Appl Microbiol Biotechnol, 2008, 78(4): 621−625.
[30] Yu YJ, Wu SC, Chan HH, et al. Overproduction of soluble recombinant transglutaminase from Streptomyces netropsis in Escherichia coli. Appl Microbiol Biotechnol, 2008, 81(3): 523−532.
[31] Yang HL, Pan L, Lin Y. Purification and on-column activation of a recombinant histidine-tagged pro-transglutaminase after soluble expression in Escherichia coli. Biosci Biotechnol Biochem, 2009, 73(11): 2531−2534.
[32] Zhao X, Shaw AC, Wang JH, et al. A novel high-throughput screening method for microbial transglutaminases with high specificity toward Gln141 of human growth hormone. J Biomol Screen, 2010, 15(2): 206−212.
[33] Noda S, Ito Y, Shimizu N, et al. Over-production of various secretory-form proteins in Streptomyces lividans. Protein Expr Purif, 2010, 73(2): 198−202.
[34] Yokoyama KI, Kunio O, Ohtsuka T, et al. In vitro refolding process of urea-denatured microbial transglutaminase without pro-peptide sequence. Protein Expr Purif, 2002, 26(2): 329−335.
[35] Zotzel J, Keller P, Fuchsbauer HL. Transglutaminase from Streptomyces mobaraensis is activated by an endogenous metalloprotease. Eur J Biochem, 2003, 270(15): 3214−3222.
[36] Marx CK, Hertel TC, Pietzsch M. Purification and activation of a recombinant histidine-tagged pro-transglutaminase after soluble expression in Escherichia coli and partial characterization of the active enzyme. Enzyme Microb Technol, 2008, 42(7): 568−575.
[37] Yokoyama KI, Utsumi H, Nakamura T, et al. Screening for improved activity of a transglutaminase from Streptomyces mobaraensis created by a novel rational mutagenesis and random mutagenesis. Appl Microbiol Biotechnol, 2010, 87(6): 2087−2096.
[38] Shimba N, Yokoyama YI, Suzuki EI. Nmr-based screening method for transglutaminases: rapid analysis of their substrate specificities and reaction rates. J Agri Food Chem, 2002, 50(6): 1330−1334.
[39] Shimba N, Shinohara M, Yokoyama KI, et al. Enhancement of transglutaminase activity by NMR identification of its flexible residues affecting the active site. FEBS Lett, 2002, 517(1/3): 175−179.
[40] Marx CK, Hertel TC, Pietzsch M. Random mutagenesis of a recombinant microbial transglutaminase for the generation of thermostable and heat-sensitive variants. J Biotechnol, 2008, 136(3/4): 156−162.
[41] Kashiwagi T, Yokoyama KI, Ishikawa K, et al. Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense. J Biol Chem, 2002, 277(46): 44252−44260.
[42] Tagami U, Shimba N, Nakamura M, et al. Substrate specificity of microbial transglutaminase as revealed by three-dimensional docking simulation and mutagenesis. Protein Eng Des Sel, 2009, 22(12): 747−752.
[43] Yang MT, Chang CH, Wang JM, et al. Crystal structure and inhibition studies of transglutaminase from Streptomyces mobaraense. J Biol Chem, 2011, 286(9): 7301−7307.
[44] Ohtsuka T, Ota M, Nio N, et al. Comparison of substrate specificities of transglutaminases using synthetic peptides as acyl donors. Biosci Biotechnol Biochem, 2000, 64(12): 2608−2613.
[45] Sugimura Y, Yokoyama KI, Nio N, et al. Identification of preferred substrate sequences of microbial transglutaminase from Streptomyces mobaraensis using a phage-displayed peptide library. Arch Biochem Biophys, 2008, 477(2): 379−383.
[46] Ohtsuka T, Sawa A, Kawabata R, et al. Substrate specificities of microbial transglutaminase for primary amines. J Agri Food Chem, 2000, 48(12): 6230−6233.
[47] Kulik C, Heine E, Weichold O, et al. Synthetic substrates as amine donors and acceptors in microbial transglutaminase-catalysed reactions. J Mol Catal B Enzym, 2009, 57(1/4): 237−241.
[48] Bommarius AS, Blum JK, Abrahamson MJ. Status of protein engineering for biocatalysts: how to design an industrially useful biocatalyst. Curr Opin Chem Biol, 2011, 15(2): 194−200.
Progress in expression and molecular modification of microbial transglutaminase
Song Liu, Dongxu Zhang, Guocheng Du, and Jian Chen
Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, China
Microbial transglutaminase, which could catalyze the cross-linking of many proteins or non-protein materials, has been widely used in food, pharmaceutical and textile industry. To enhance the yield of the enzyme and establish corresponding platform for molecular modification, the researchers of Japanese Ajinomoto began to construct the recombinant strain producing transglutaminase in the 1990s. So far, the enzyme has been successfully expressed in different expression systems. Some of the recombinant strains are more productive than wild strains. Recently, progress has been made in the molecular modification of microbial transglutaminase, and the activity, thermo-stability and specificity of the enzyme are improved. This review briefly summarized and analyzed the strategies involved in these studies, and noted its trends.
Streptomyces, transglutaminase, protein expression, molecular modification, protein structure
谷氨酰胺转胺酶 (Transglutaminase,EC 2.3.2.13, TGase),能够催化蛋白质肽链中谷氨酰胺残基的 γ-羧酰胺基与各种酰基受体发生酰胺基转移反应,形成 ε-(γ-谷氨酰基) 赖氨酸共价键。根据酰基受体不同,TGase催化反应分3类:1) 交联反应:当蛋白质中赖氨酸残基的ε-氨基作为酰基受体时,催化形成的ε-(γ-谷氨酰基) 赖氨酸共价键导致蛋白质分子内或分子间发生交联,形成网络结构 (图1a);2) 连接反应:当伯胺作为酰基受体时,催化形成的 ε-(γ-谷氨酰基)赖氨酸共价键使游离的伯胺与蛋白质连接 (图1b);3)脱酰胺:当不存在伯胺时,水成为酰基受体,TGase催化蛋白质中谷氨酰胺残基的γ-羧酰胺基脱去氨基生成谷氨酸残基 (图1c)[1-2]。
May 4, 2011; Accepted: September 16, 2011
Supported by:National Natural Science Foundation of China (Nos. 31000031, 31171639), National High Technology Research and Development Program of China (863 Program) (No. 2011AA100905), Natural Science Foundation of Jiangsu Province (No. BK2010147).
Guocheng Du. Tel/Fax: +86-510-85918309; E-mail: gcdu@jiangnan.edu.cn
国家自然科学基金 (Nos. 31000031, 31171639),国家高技术研究发展计划 (863计划) (No. 2011AA100905),江苏省自然科学基金 (No. BK2010147) 资助。
