阳离子胶束介质中次磷酸钠还原偶氮胂I褪色催化动力学光度法测定痕量硒(IV)
2010-12-02周之荣王群章淑媛
周之荣,王群,章淑媛
(1.广东药学院公共卫生学院,广东 广州 510240;2.东华理工大学校医院,江西 抚州 344000)
硒是必需微量营养元素,硒的缺乏和过剩都与人的生命休戚相关。适量的硒具有抗癌和改善心肌等功能;人体缺硒易引起克山病、大骨节病、白肌病等;而过高的硒含量将引起食欲减退、肝脏受损、毛发改变等病症。硒在植物(十字花科)的生长周期中起着重要作用,半干旱地区的植物可以吸收富集在土壤中的有机硒化合物有可能对当地的牲畜造成危害。从含硒土壤渗出和通过工业废水的排放进入自然水体的硒将带来严重的环境污染和生物毒性[1]。
随着科学技术的发展和人类生活水平的提高,人们越来越重视微量元素硒与人类生活的关系,硒的分析方法近年来也有了较大的发展。测定硒的分析方法很多,大部分是分光光度法[2]。但存在费时、处理过程复杂、使用有毒试剂和测定体系不太稳定等不足。氢化物发生原子吸收光谱法[3]和氢化物发生原子荧光光谱法[4]也是测定痕量硒的灵敏方法,但存在共存离子干扰。溶出伏安法[5]、中子活化法[6]、毛细管电泳[7]、气相色谱法[8]、高效液相色谱法[9]、X 射线荧光光谱法[10]、化学发光淬灭分析法[11]、催化极谱法[12]以及可以在线检测的电感偶合等离子体原子发射光谱法[13]、电感耦合等离子体质谱法[14]、亦可以用于测定痕量硒。其中一些方法具有较好的灵敏度,但需要昂贵的试剂和仪器;有的还需要预先富集才能测定。因此有必要建立灵敏而准确地定量测定痕量硒的新方法。
催化动力学方法具有仪器价廉、灵敏度高、操作简便、分析成本低、便于推广等特点,倍受国内外学者关注,在痕量硒的测定中已有很多报道[15-21]。由于动力学反应的特殊性以及报道方法的灵敏度或选择性等方面的限制,建立新的测定体系仍具有一定的实际意义。
1 材料与方法
1.1 仪器与主要试剂
U-3010型分光光度计:日本日立公司;721-W型分光光度计:上海分析仪器厂;501型超级恒温器:上海实验仪器总厂。
硒(IV)标准溶液:1.0 g/L标准储备液(1.0 mol/L HCl介质),用时逐级稀释至0.01 mg/L的工作液;次磷酸钠溶液0.05 mol/L,使用当天配制并装入棕色瓶中;偶氮胂 I(AsA I)溶液:0.02 mol/L;H2SO4溶液:0.5 mol/L,0.1 mol/L。
氯化十六烷基吡啶(CPC)、Triton X-100、乳化剂OP、十二烷基磺酸钠(SDS)、溴化十六烷基三甲基铵(CTMAB)溶液:1.2×10-2mol/L。
所用试剂均为分析纯,试验用水为二次石英亚沸水。
1.2 方法
取两支刻度一致的50 mL具塞比色管,向其中一支加入0.02μg硒(IV)标准溶液,另一支不加(非催化反应),水稀释至10 mL。分别加入1滴1 g/L的2,4-二硝基苯酚溶液,用7.0 mol/L NH3·H2O调至黄色,再以0.1mol/L H2SO4调至黄色刚消失。加入0.50mol/L H2SO4溶液1.0 mL,1.2×10-2mol/L CPC 溶液 1.0 mL,0.05 mol/L次磷酸钠溶液1.0 mL,0.02 mol/L AsA I溶液1.0 mL。用水稀释至25 mL,摇匀。半启玻塞,置于90℃水浴中加热9 min(秒表计时)。迅速取出流水中冷却5 min后,以水为参比,用1 cm比色皿,在波长500 nm处测量吸光度A0(空白)和A(试液),计算ΔA(=A0-A)。
2 结果与讨论
2.1 吸收曲线

配制图1所示的不同组分溶液,按试验方法操作,在U-3010型分光光度计上扫描并绘制吸收曲线。结果表明:在酸性介质中存在胶束介质时,次磷酸钠能还原AsA I褪色,但褪色速率较慢。而痕量硒(IV)加入能明显的催化次磷酸钠还原AsA I的褪色反应,且催化褪色程度与所加入的硒(IV)量在一定范围内成正比。所有曲线在500 nm处均有最大吸收值,故试验选择测定波长为500 nm。
2.2 反应介质的选择及用量影响
试验了稀 H2SO4、HAc、HNO3、HCl、H3PO4及NaAc-HAc缓冲溶液介质对反应体系的影响。结果表明,在稀 HNO3、HAc、HCl、H3PO4及 NaAc-HAc 缓冲溶液介质中,催化反应和非催化反应无明显区别,在稀硫酸介质中硒(IV)具有明显的催化作用,故本文选用稀硫酸为反应介质。反应体系中0.50 mol/L H2SO4溶液的加入量在0.8 mL~1.2 mL时,ΔA值最大且稳定。试验选用0.50 mol/L的H2SO4溶液1.0 mL,此时体系酸度为0.02 mol/L 的 H2SO4。
2.3 表面活性剂的种类和用量的影响
为选择适当的胶束介质加速反应,必须考虑反应物种的类型和电荷,因为胶束的加速影响必须存在反应和胶束表面之间的静电及亲水相互反应。试验了同样浓度(均超过各自的临界胶束浓度(c.m.c.))的阳离子表面活性剂(CPC,CTMAB)、阴离子表面活性剂(SDS)、非离子表面活性剂(Triton X-100,乳化剂 OP)对催化反应的影响,结果见表1。
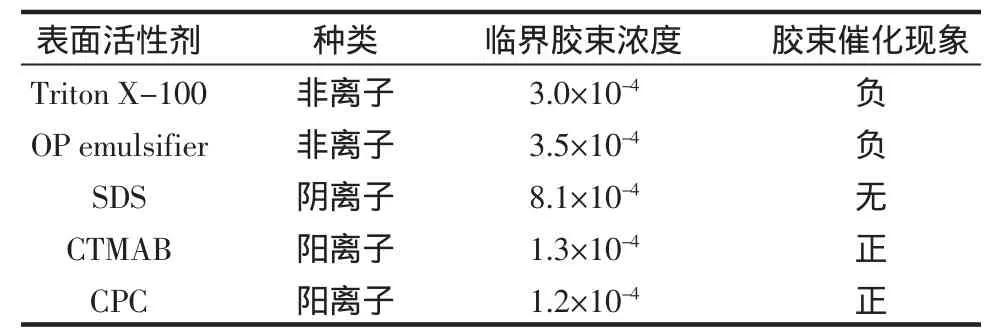
表1 表面活性剂对加速Se(IV)-AsA I-NaH2PO2反应体系的影响Table 1 Surfatants tested as potential micellar catalysts for the enhanced rate of Se(IV)-AsA I-NaH2PO2reaction system
Se(IV)和H2PO2-带负电荷,AsA I在测定的pH时可能带负电荷或不带电荷,这样可以推测阳离子表面活性剂可以加快Se(IV)AsA I NaH2PO2反应体系的速度是可行的。事实上阳离子表面活性剂CPC、CTMAB能够增加体系的灵敏度,而CPC的效果更好,故选择CPC进行进一步试验。
2.4 试剂用量的影响
2.4.1 次磷酸钠用量的影响
当0.05 mol/L NaH2PO2用量在0.8 mL~1.2 mL时,ΔA值最大且相对变化较小,此时的灵敏度较高。故试验选用1.0 mL。
2.4.2 AsA I用量的影响
AsA I用量在0.5 mL~1.0 mL时,ΔA随AsA I用量的增加而增大,但同时非催化反应体系的吸光度亦明显增大,并在AsA I用量为1.0 mL时达到最大;超过1.0 mL后,ΔA值反而减少。故试验选用0.02 mol/LAsAI溶液1.0 mL。
2.4.3 CPC浓度及用量的影响
试验了CPC浓度在2.4×10-4mol/L~1.2×10-3mol/L(用量0.5 mL~2.5 mL)时对反应速率的影响。结果表明,ΔA随CPC浓度的增加而增大,但同时非催化反应体系的吸光度亦明显增大,并在CPC浓度为4.8×10-4mol/L时达到最大;超过1.0 mL后,ΔA值反而减少,可能是超过一定浓度后表面活性剂的聚集和溶液中AsA I摩尔吸光度的改变。故试验选用CPC浓度为4.8×10-4mol/L(用量 1.0 mL)。
2.5 反应温度对反应速率的影响
在室温下,反应几乎不发生,故可以用流水冷却来终止反应;在40℃~60℃时,ΔA随着温度的升高而缓慢增加,但催化反应亦不明显;超过60℃以上时,随着温度的升高ΔA明显增加。在70℃~90℃时,催化反应明显加快,ΔA明显增加,并在90℃时达到最大。超过90℃时,非催化反应也明显加快,ΔA值反而降低。为便于操作和获得较高灵敏度,本试验在90℃水浴中加热。
2.6 加热时间的影响
试验结果表明:在3 min~8 min内ΔA随反应时间的加长而增大,且呈线性关系,其线性方程为:ΔA=0.0649 t-0.1081(r=0.9992)。反应的速率常数 k0=1.2×10-3/s。本文选用加热反应时间为8 min,取出用流水冷却5 min终止反应后,体系吸光度至少2 h保持不变。
2.7 校准曲线
试验结果表明,Se(IV)量在 0~1.0μg/L 范围内服从比尔定律。其线性回归方程:ΔA=4.268 ρSe(IV)(μg/25 mL)-1.298×10-3。式中 ρSe(IV)为 25 mL 体积中 Se(IV)的微克数,相关系数为0.9992。按试验方法,对0.02、0.01μg/25 mL Se(IV)的试液进行 11 次测定,相对标准偏差分别为1.9%和2.1%;对空白进行11次测定,标准偏差为1.90×10-3。由空白溶液的3倍标准偏差除以工作曲线的斜率求出检出限为0.053μg/L。
2.8 共存离子的影响
对于测定25 mL溶液中的0.02μg Se(IV),相对误差≤±5%时,共存离子的允许量(μg)为结果见表2。当共存离子超过允许量后,按文献[15]利用巯基葡聚糖胶进行分离。

表2 共存离子的影响Table 2 Interference study for the determination of Se(IV)at the optimum reagent concentration
2.9 动力学参数的测定
2.9.1 反应级数的测定
催化动力学测定Ti(IV)的化学反应总方程式可以表示如下:
AsA I+硫酸+次磷酸钠+Se(IV)→反应产物
该反应的速率方程式可表示为:

式中:R为反应速率,[mol/(L·min)];C为反应物浓度,(mol/L);α,β,γ,δ为反应级数。用单因数变换固定时间法测定各反应物的反应级数,结果如下:

由直线方程的斜率可知:α=1.0213≈1,β=0.9878≈1,γ=1.042≈1,δ=0.087≈0,故本催化反应对 Se(IV)、AsA I和次磷酸钠而言为一级反应,对硫酸而言为零级反应。由此可见,在稀硫酸介质中,以Se(IV)为催化剂,次磷酸钠还原AsA I褪色反应的速率方程式可表示为:

在试验条件下,c硫酸,c次磷酸钠,cAsAI垌ρSe(IV),且基本保持不变,速率方程可以简化为:

将上式积分得:-cAsAI=k2ρSe(IV)t
代入 A=εbcAsAI中,得 A=-εbk2ρSe(IV)t,则:ΔA=A0-A=εbk2ρSe(IV)t
采用固定时间法测定吸光度,动力学方程为:ΔA=k3ρSe(IV)
即ΔA与催化剂Se(IV)在一定浓度范围内成正比,这就是本催化反应测定痕量Se(IV)的定量依据。
2.9.2 表观活化能的测定
固定反应物浓度和加热时间,在80℃~90℃时,测定催化反应的吸光度A和非催化反应吸光度A0,计算 ΔA=A0-A,作-lgΔA~(1/T)×103曲线。结果表明,-lgΔA与(1/T)×103存在着线性关系,用最小二乘法处理,其线性回归方程为:-lgΔA=2.386×(1/T)×103-5.824(r=0.9972),根据Arrhenius公式计算表观活化能为:Ea=2.386×2.303×8.314=45.69 kJ/mol。
3 样品分析
称取1.0000 g样品于聚四氟乙烯坩埚中,加入3.0 mL浓硝酸和1.0 mL 30%过氧化氢,再放入高压溶解罐中,旋紧罐盖,放入微波炉中。分别在100℃、150℃下溶解1 h和3 h。冷却至室温后,取出聚四氟乙烯坩埚,将消化后的样品转移到烧杯中。再加热至近干,除去过量的硝酸和过氧化氢,用适量的4.5 mol/L HCl浸取Se(IV)。加入NaOAc调节离子强度,使其浓度为0.1 mol/L,再用HCl调节酸度为4.5 mol/L HCl后,以5 mL/min的流速通过SDG吸附柱,只有Ag+和Se(IV)被吸附。再用3.0 mL HCl和2.0过氧化氢洗脱柱上的Se(IV)[15],加热除去过量的过氧化氢后,转移到100 mL容量瓶中,用水定容。取适量溶液按试验方法测定,并与ICP-AES法对照,结果相符。测定结果见表3。

表3 样品中Se(IV)的测定结果和回收率Table 3 Analysis results of Se(IV)and recoveries in samples
4 结论
本文研究在0.02 mol/LH2SO4介质中,痕量硒(IV)对次磷酸钠还原偶氮胂I(AsA I)的褪色反应有明显的催化作用,将阳离子表面活性剂氯化十六烷基吡啶(CPC)用于增加 Se(IV)-AsA I-NaH2PO2反应体系的灵敏度和稳定性。研究了适宜的动力学反应条件,结合巯基葡聚糖凝胶分离富集,建立了测定痕量硒(IV)的催化动力学光度法,已用于测定食品和人发样品中的痕量硒(IV)。方法检出限为0.053μg/L,线性范围为 0.0~1.0μg/L。
[1]Standard methods for the examination of water and wastewater[M].19th ed.Washington,D C:American Public Health Association Press,1995:3-85
[2]Shlyapunova E V,Sergeeva V P,Sergeev G M.Highly sensitive redox-photometric determination of selenite and iodide ions in mineral waters[J].Journal of Analytical Chemistry,2008,63(3):219-222
[3]Maleki N,Safavi A,Doroodmand M M.Determination of selenium in water and soil by hydride generation atomic absorption spectrometry using solid reagents[J].Talanta,2005,66(4):858-862
[4]Fan H F,Wen H J,Hu R Z,et al.Determination of total selenium in geological samples by HG-AFS after concentration with thiol cotton fiber[J].Chinese Journal of Geochemistry,2008,27(1):90-96
[5]Stozhko N Y,Morosanova E I,Kolyadina L I,et al.Ceramic composite electrode for the determination of selenium (IV) by stripping voltammetry[J].Journal of Analytical Chemistry,2006,61(2):158-165
[6]Chajduk E,Polkowska-Motrenko H,Dybczyński R S.A definitive RNAA method for determination of selenium in biological samples:uncertainty evaluation and assessment of degree of accuracy[J].Accreditation and Quality Assurance:Journal for Quality,Comparability and Reliability in Chemical Measurement,2008,13(8):443-451
[7]Dzierzgowska M,Pyrzy ska K,Pobozy E.Capillary electrophoretic determination of inorganic selenium species [J].Journalof Chromatography A,2003,984(2):291-295
[8]Gómez-Ariza J L,Pozas J A,Giráldez I,et al.Comparison of three derivatization reagents for the analysis of Se (IV) based on piazselenol formation and gas chromatography-mass spectrometry[J],Talanta,1999,49(2):285-292
[9]Mazej D,Falnoga I,Veber M,et al.Determination of selenium species in plant leaves by HPLC-UV-HG-AFS[J].Talanta,2006,68(3):558-568
[10]Gomes A C F,Menegario A A,Pellegrinotti D C,et al.A hydride generation flow system for determination of arsenic and selenium by total reflection X-ray fluorescence spectrometry[J].Spectrochimica Acta Part B:Atomic Spectroscopy,2004,59(9):1481-1484
[11]易钢,颜玉蓉,钟梁.静态化学发光淬灭分析法测定血清硒[J].重庆医科大学学报(Chinese Journal of Chongqing Medical University),2006,31(3):119-120,128
[12]孙莉,李方实.催化极谱法测定生物样品中的微量硒[J].南京工业大学学报:自然科学版,(Chinese Journal of Nanjing University of Technology(Natural Science Edition)),2006,28(3):82-85
[13]Masson P,Orignac D,Prunet T.Optimization of selenium determination in plant samples by hydride generation and axial view inductively coupled plasma atomic emission spectrometry[J].Analytica Chimica Acta,2005,545(1):79-84
[14]Kobayashi K,Katsuya Y,Abdulah R,et al.Rapid and direct determination of selenium,copper,and zinc in blood plasma by flow injection-inductively coupled plasma-mass spectrometry[J].Biological Trace Element Research,2007,115(1):87-93
[15]Zhou Z R,Wang Q,Zhang S Y,et al.Catalytic kinetic spectrophotometric determination of trace selenium (IV)in microemulsion after separation and enrichment by SDG[J].Chinese Journal of Food Science,2008,29(6):292-297
[16]Gudzenko L V,Pantaler R P,Blank A B.Catalytic spectrophotometric determination of nanogram amounts of selenium(IV)[J].Journal of Analytical Chemistry,2004,59(10):935-938
[17]Gong Z J,Zhang X S,Chen G H,et al.Flow injection kinetic spectrophotometric determination of trace amounts of Se(IV)in seawater[J].Talanta,2005,66(4):1012-1017
[18]Parham H,Jafarpoor J.Kinetic-spectrophotometric determination of trace amounts of selenium (IV)based on its catalytic effect on the reduction of brilliant green by sulphide ion[J].Indian Journal of Chemistry Section a-Inorganic Bio-Inorganic Physical Theoretical&Analytical Chemistry,2001,40(12):1362-1364
[19]Gurkan R,Akcay M.Kinetic spectrophotometric determination of trace amounts of selenium based on the catalytic reduction of maxilon blue-SG by sulfide[J].Microchemical Journal,2003,75(1):39-49
[20]Ensafi A A,Lemraski M S.Highly sensitive spectrophotometric reaction rate method for the determination of selenium based on the catalytic reduction of sulfonazo by sulfide[J].Analytical Letters,2004,37(12):2469-2483
[21]Khajehsharifi H,Mousavi M F,Ghasemi J,et al.Kinetic spectrophotometric method for determination of selenium and tellurium using partial least squares calibration[J].Analytica Chimica Acta,2004,512(2):369-373
[22]Rubio S,Pérez-Bendito D.Surfactant to dye binding degree based approach for the selective determination of L-glutamate in foodstuffs[J].Anal.Chem.Acta.,1989,224(2):85-89
