外消旋联萘二酸合成方法的改进
2010-11-26叶康曹锰刘丽琴黄巍张万轩
叶康,曹锰,刘丽琴,黄巍,张万轩
(湖北大学 有机功能分子合成与应用教育部重点实验室,湖北 武汉 430062)
外消旋联萘二酸1是有机合成中非常重要的中间体.可用马钱子碱或者奎宁拆分得到光学活性的(R)-联萘二酸和(S)-联萘二酸.Maruoka[1-2]小组利用光学活性的联萘二酸作中间体,合成出许多基于联萘结构的手性季胺盐,并广泛应用在不对称催化中.关于外消旋联萘二酸的合成已有文献报道[3-5].Colletti和Halterman[3]等人用碳酸钙作弱碱,对中间体1-溴-2-溴甲基萘4进行水解反应,生成1-溴-2-羟甲基萘5.碳酸钙在水中溶解度较小,反应体系为两相,反应重复性不好;Weber[4]将1-溴-2-溴甲基萘4与六次甲基四胺,醋酸和盐酸反应生成1-溴-2-萘甲醛,再用高锰酸钾将1-溴-2-萘甲醛氧化成1-溴-2-萘甲酸6,反应总产率较低,不经济.
本文中以Colletti和Halterman的路线为基础,以β-甲基萘2为原料经7步反应合成了联萘二酸(如Scheme 1),对其中1-溴-2-羟甲基萘5的合成进行了改进:先用醋酸钾进行取代反应,再用氢氧化钠溶液水解,避免了分子间反应生成醚类,改进后的操作更加简单,重复性好,产率高达92%;此外,文中对Ullmann偶联法合成1,1′-联萘-2,2′-二甲酸甲酯8的方法以及催化剂活性铜粉的制备也进行了详细地探讨. 化合物1~8的结构如Scheme 1所示(下同).
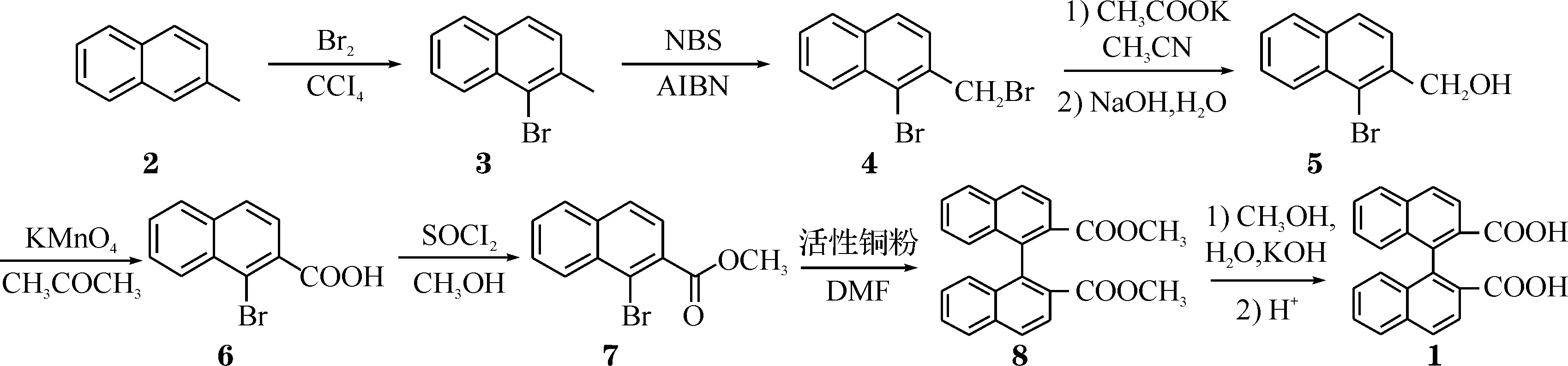
Scheme 1 联萘二酸的合成路径
1 实验部分
1.1仪器与试剂及处理市售DMF(N,N′-二甲基甲酰氨)用活化(300 ℃)的4A分子筛干燥后重蒸,重蒸后的DMF放入活化的分子筛保存备用;NBS(溴代琥珀酰亚胺)和AIBN(偶氮二异丁腈)经重结晶提纯;活性铜粉是用锌粉置换硫酸铜得到湿润状态下的铜粉[6],经过碘的丙酮溶液处理[7],然后真空干燥(用P2O5作干燥剂).温度计未经校正;红外光谱仪:PE-Spectrum One 型红外光谱仪,KBr压片;核磁共振仪:INOVA(600 MHz)核磁共振仪,以CDCl3或DMSO-d6为溶剂,TMS(四甲基硅)为内标.
1.2化合物3的合成[8]在一个装有机械搅拌器的500 mL三颈烧瓶中,加入化合物2(71 g,500 mmol)的CCl4(150 mL)溶液,并加入碘(少许)和还原铁粉(少许).混合物冰浴冷却到0 ℃.在一恒压滴液漏斗中,将液溴(80 g,500 mmol)溶解于CCl4(150 mL).在避光条件下,边搅拌边滴入液溴溶液,8 h之内滴加完毕.滴加过程中,冰浴温度不能超过5 ℃,滴加完后放置过夜.混合物依次用10% NaOH溶液和水洗涤,有机相用无水MgSO4干燥,过滤,减压(5 mmHg)蒸馏收集132.5~133.5 ℃外产物3(91.75 g),(产率83%).1H NMR(600 MHz,CDCl3,ppm):δ=8.28(1H,d,Ar),7.76(1H,d,Ar),7.67(1H,d,Ar),7.53~7.56(1H,m,Ar),7.43~7.45(1H,m,Ar),7.32(1H,d,Ar),2.61(3H,s,Ar).
1.3化合物4的合成将1-溴-2-甲基萘3(30 mL,195 mmol)溶解在无水CCl4(245 mL)中,在稳定的氩气氛中,加入NBS(36.67 g, 206 mmol)和AIBN(84 mg).反应混合物用100 W电灯泡照射,回流3 h,得到琥珀酰亚胺的悬浊液.趁热过滤溶液,得到的固体溶解于热水中,用热的四氯化碳萃取,合并有机相和滤液,浓缩,得到白色晶体二溴化物4(50.89 g),产率87%.mp:104~106 ℃(lit.[3]100 ℃;lit.[4]107~109 ℃);1H NMR(600 MHz,CDCl3,ppm):δ=8.33(1H,d,Ar),7.79~7.82(2H,m,Ar),7.51~7.63(3H,m,Ar),4.86(2H,s,CH2);IR(KBr,cm-1):v=1 499,1 328,1 204,978,809,755,670.
1.4化合物5的合成在搅拌下,向1-溴-2-溴甲基萘4(8.61 g,29 mmol)的乙腈(120 mL)溶液中,加入乙酸钾(10 g,102 mmol)和溴化四丁基胺(0.28 g,3 mol%),回流4~5 h.TLC检测原料已经消耗完后,加入2 mol/L NaOH溶液100 mL,继续加热4 h.蒸去乙腈,用水和二氯甲烷萃取,合并有机相,用无水MgSO4干燥,过滤,重结晶,得到白色针状晶体5(6.33 g),产率92%.mp:98~99 ℃(lit.[3]100 ℃;lit.[4]101~102 ℃);1H NMR(600 MHz,CDCl3,ppm):δ=8.32(1H,d,Ar),7.83~7.85(2H,m,Ar),7.52~7.64(3H,m,Ar),5.00(2H,d,CH2),2.11~2.13(1H,m,OH);IR(KBr,cm-1):v=3 231,2 892,1 502,1 461,1 062,963,860,808,763,735,653.
1.5化合物6的合成将1-溴-2-羟甲基萘5(8.20 g,34 mmol)溶解到丙酮(50 mL)中,加热至回流状态,然后将高锰酸钾(17.6 g,100 mmol)溶解到丙酮和水(200 mL/40 mL)的混合溶剂中,缓慢加热,并逐滴滴入到1-溴-2-羟甲基萘5中,30 min之内滴完.反应混合物加热回流5 h,冷却到室温,通过硅藻土过滤,并用无水乙醚洗涤,旋蒸除去溶剂.向混合物中加入乙醚和2 mol/L NaOH溶液,萃取分离.在冰水浴条件下,水相用2 mol/L HCl小心中和,产生白色固体,用乙醚萃取,有机相用无水MgSO4干燥,过滤,去溶剂,得到白色固体6(5.98 g),产率70%.mp:181~182 ℃(lit.[3-4]108~109 ℃);1H NMR(600 MHz,DMSO-d6,ppm):δ=8.28(1H,d,Ar),7.97~8.01(2H,m,Ar),7.58~7.73(3H,m,Ar),3.39(br,1H,COOH); IR(KBr,cm-1):v=3 421,1 698,1 463,1 402,1 279,1 250,972,761.
1.6化合物7的合成将1-溴-2-萘甲酸6(10.09 g,40 mmol)溶解到甲苯(25 mL)中,向溶液中加入二氯亚砜(25 mL),混合物加热到80 ℃,搅拌回流3 h,蒸去溶剂(依次用水泵,油泵抽真空),得到黄色固体.将此固体溶解到无水甲醇中(80 mL),搅拌至全部溶解,加热回流5 h,反应结束之后,将无水甲醇旋蒸除去,用石油醚重结晶,得到白色固体7(9.76 g),产率92%.mp:57~59 ℃(lit.[3]58 ℃; lit.[4]53~55 ℃);1H NMR(600 MHz,CDCl3,ppm):δ=8.46(d,1H,Ar),7.83~7.86(m,2H,Ar),7.66~7.69(m,2H,Ar),7.60~7.62(m,1H,Ar),4.00(s,3H,CH);IR(KBr,cm-1):v=2 948,1 720,1 458,1 266,1 237,1 126,1 002,761.
1.7化合物8的合成将新制的活化并干燥的铜粉(1.33 g,21 mmol)加入到一个烘干带有冷凝管的烧瓶中.1-溴-2-萘甲酸甲酯7(0.68 g,5.6 mmol)溶解于DMF(10 mL),加入到活性铜粉中.160 ℃条件下加热6 h,TLC检测反应结束之后,混合物通过硅藻土过滤,并用热的甲苯洗涤.有机相依次用2 mol/LHCl,H2O和盐水萃取洗涤,用无水MgSO4干燥,过滤,蒸干溶剂,剩余物经柱层析分离(乙酸乙酯:石油醚=1∶10)得到白色固体8(1.51 g),产率73%.mp:153~154 ℃(lit.[3]154 ℃;lit.[4]157~158 ℃);1H NMR(600 MHz,CDCl3,ppm):δ=8.18(d,2H,Ar),8.01(d,2H,Ar),7.94(d,2H,Ar),7.50~7.53(m,2H,Ar),7.22~7.25(m,2H,Ar),7.07(d,2H,Ar),3.49(s,6H,CH);IR(KBr,cm-1):v=3 056,2 948,1 723,1 621,1 596,1 458,1 429,1 281,1 243,1 189,1 135,1 065,833,767.
1.8化合物1的合成将1,1′-联萘-2,2′-二甲酸甲酯8(1.6 g,4.3 mmol)加入到热的KOH(8.0 g,143 mmol)的甲醇(40 mL)溶液中,反应回流15 h,蒸去溶剂,向其中加入40 mL蒸馏水,碱性的水溶液冷却到0 ℃,并用浓HCl中和.待有机二酸沉淀之后,产物抽滤并用水洗涤,真空干燥得到白色晶体1(1.38 g),产率94%.1H NMR(600 MHz,DMSO-d6,ppm):δ=12.38(b,2H,OH),8.04~8.09(m,6H,Ar),7.54~7.56(m,2H,Ar),7.27~7.30(m,2H,Ar),6.88~6.89(m,2H,Ar); IR(KBr,cm-1):v=2 994,1 689,1 620,1 597,1 466,1 411,1 285,1 254,1 170,829,766.
2 结果与讨论
在1-溴-2-溴甲基萘4的水解反应中,与亚甲基相连的溴原子在碱性条件下离去,形成碳正离子.如果直接用NaOH溶液水解,会发生分子间反应生成醚类,因此通常在弱碱性条件下水解.Colletti和Halterman的合成路线中,用CaCO3和H2O的悬浊液作为反应试剂,因CaCO3在水中的溶解度较小,反应体系为两相,该步反应的重复性不好.我们对此作了改进,用醋酸钾的乙腈溶液与4反应,加入少量n-Bu4NBr作为相转移催化剂,先生成酯9(如Scheme 2),待亲核取代反应结束之后,再用NaOH溶液水解得到醇5,两步产率可以达到92%.改变方法之后,虽然为两步反应,但是采用“一锅法”,中间不需要进行分离,直接加入NaOH水溶液加热即可,这样操作简单,还可以避免醚的生成,重复性也很好.
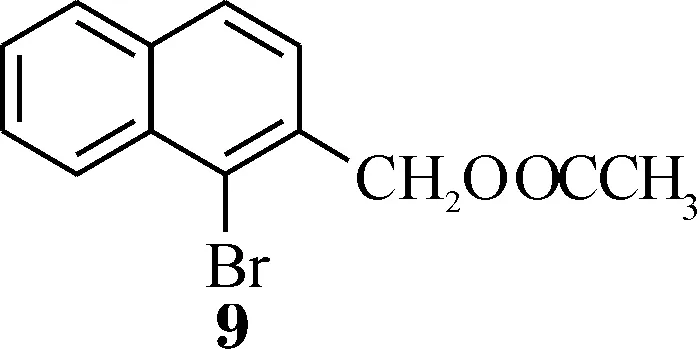
Scheme 2 乙酸-β-溴萘基甲酯的化学式
此外,对制备8的Ullmann偶联反应及铜催化剂的制备进行了详细的研究.在Ullmann偶联反应中,活性铜粉的活性和干燥程度对于反应产率是至关重要的[9],痕量水存在时,原料虽然基本消耗完,但反应会得到大量还原产物β-萘甲酸甲酯,导致产物8的产率大幅度降低,若铜粉活性不够则会有大量原料不能反应.但活性铜粉的干燥程度及杂质的含量难以直观的判断.在本实验中,按照文献方法[8],将五水硫酸铜溶解于热水中,冷却至室温,搅拌下分批加入过量锌粉.析出的铜粉用水以倾析法洗涤,并加入稀盐酸以除去过量的锌粉,继续激烈搅拌到无氢气逸出后过滤,水洗,得到湿润状态的铜粉用2%碘的丙酮溶液浸泡10 min(以使铜粉充分活化),过滤,用稀盐酸和丙酮混合液洗涤,最后用丙酮洗涤3次,然后真空干燥(用P2O5作干燥剂,P2O5必须是粉末状态以保证良好的干燥效果),干燥时间48 h以上,得到的活性铜粉可以有效地催化偶联反应,产率由30%提高到70%.应当注意的是,在实验操作过程中,实验装置必须彻底除去水和空气.
参考文献:
[1] Kitamura M, Shirakawa S, Maruoka K.Powerful chiral phase-transfer catalysts for the asymmetric synthesis of α-alkyl- and α,α-dialkyl-α-amino acids[J].Angew Chem Int Ed, 2005,44:1549-1551.
[2] Ooi T, Uematsu Y, Maruoka K.New improved procedure for the synthesis of structurally diverse N-spiro C2- symmetric chiral quaternary ammonium bromides[J].J Org Chem, 2003,68:4576-4578.
[3] Colletti S L,Halterman R L.Asymmetric synthesis and metalation of binaphthyl cyclopentadiene, a C2-symmetric chiral cyclopentadiene[J].Organometallics, 1991,10:3438-3448.
[4] Weber E, Csoregh I, Stensland B, et al.A novel clathrate design:selective inclusion of uncharged molecules via the binaphthyl hinge and appended coordinating groups. X-ray crystal structures and binding modes of 1,1′-binaphthyl-2,2′-dicarboxylic acid host/hydroxylic guest inclusions[J].J Am Chem Soc,1984,106:3297-3306.
[5] Seki M, Yamada S, Kuroda T, et al.A practical synthesis of C2-symmetric chiral binaphthyl ketone catalyst[J].Synthesis, 2000, 12:1677-1680.
[6] 黄枢,谢如刚,田宝芝,等.有机合成试剂制备手册[M].2版.北京:科学出版社,2005.
[7] Kleiderer E C, Adams R.Stereochemistry of diphenylsⅩⅩⅪ′:preparation and properties of 2,2′,6,6′-tetrafluo-3,3′-dicarboxy-5,5′-dichlorodiphenyl[J].J Am Chem Soc, 1983(03):4219-4225.
[8] Adams R, Binder L O.Restricted rotation in aryl olefins(Ⅲ):preparation and resolution ofβ-chloro-β-(2-methyl-1-naphthyl)-acrylic acids[J].J Am Chem Soc,1941(10):2773-2776.
[9] Weston P E, Adkins H.Catalysis with copper in the Ullmann reaction[J].J Am Chem Soc, 1928,50(3):859-866.
