JNK1, JNK2, and JNK3 are involved in P-glycoprotein-mediated multidrug resistance of hepatocellular carcinoma cells
2010-07-05
Xiamen, China
JNK1, JNK2, and JNK3 are involved in P-glycoprotein-mediated multidrug resistance of hepatocellular carcinoma cells
Feng Yan, Xiao-Min Wang, Zhong-Chen Liu, Chao Pan, Si-Bo Yuan and Quan-Ming Ma
Xiamen, China
BACKGROUND: Multidrug resistance (MDR) is extremely common in hepatocellular carcinoma (HCC) and is a major problem in cancer eradication by limiting the efficacy of chemotherapy. Modulation of c-Jun NH2-terminal kinase (JNK) activation could be a new method to reverse MDR. However, the relationship between JNK activity and MDR in HCC cells is unknown. This study aimed to explore the relationship between MDR and JNK in HCC cell lines with different degrees of MDR.
METHODS: A MDR human HCC cell line, SMMC-7721/ ADM, was developed by exposing parental cells to gradually increasing concentrations of adriamycin. The MTT assay was used to determine drug sensitivity. Flow cytometry was used to analyze the cell cycle distribution and to measure the expression levels of P-glycoprotein (P-gp) and MDR-related protein (MRP)-1 in these cells. JNK1, JNK2 and JNK3 mRNA expression levels were quantified by real-time PCR. Expression and phosphorylation of JNK1, JNK2, and JNK3 were analyzed by Western blotting.
RESULTS: The MDR of SMMC-7721/ADM cells resistant to 0.05 mg/L adriamycin was mainly attributed to the overexpression of P-gp but not MRP1. In addition, these cells had a significant increase in percentage in the S phase, accompanied by a decrease in percentage in the G0/G1 phase, which is likely associated with a reduced ability for cell proliferation and MDR generation. We found that JNK1, JNK2, and JNK3 activities were negatively correlated with the degree of MDR in HCC cells.
CONCLUSION: This study suggests that JNK1, JNK2, and JNK3 activities are negatively correlated with the degree of MDR in HCC cells.
(Hepatobiliary Pancreat Dis Int 2010; 9: 287-295)
multidrug resistance; c-Jun NH2-terminal kinase; hepatocellular carcinoma; P-glycoprotein; multidrug resistance-associated protein
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common disease in the world and the third major cause of cancer-related deaths.[1,2]The incidence of this tumor type ranges from approximately 10 cases per 100 000 in North America and Western Europe to 50-150 per 100 000 in Asia and Africa.[3]HCC is a hypervascular solid cancer characterized by a high degree of drug resistance.[4]The development of multidrug resistance (MDR) to chemotherapeutic agents in HCC cells is the major cause for failure of cancer therapy.[5]
MDR cancer cells may appear upon prolonged treatment with anti-cancer drugs. The development of MDR poses a great obstacle in chemotherapy for cancer because higher doses of drugs need to be administered and in turn may cause severe adverse effects. Most strategies that have been developed to reverse MDR in cancer cells involve the use of resistance modulators. These modulators have the ability to reverse resistance through inhibiting the function of MDR transporters [e.g. P-glycoprotein (P-gp) and MDR-related protein (MRP)].[6,7]Another strategy to circumvent MDR could be down-regulating the expression of genes encoding these transporters. However, the regulation of MDR-related gene expression is highly complex and is embodied in multiple transcription-regulatoryelements contained in 5' and 3' flanking sequences of the MDR-1 gene as well as numerous cell-type- and stimulusdependent protein factors involved in transcriptionregulatory processes.[8]Understanding of the molecular mechanisms and signal-transduction pathways involved in the regulation of MDR-related genes could be useful in future to overcome MDR and improve chemotherapeutic efficacy.
The c-Jun NH2-terminal kinase (JNK) pathway is known to play critical roles in diverse cellular processes. JNK is activated when cells are exposed to proinflammatory cytokines, environmental stress (e.g. UV radiation, heat shock, osmotic shock, and redox stress), and various anticancer drugs, or when undergoing growth factor withdrawal. The role of the JNK pathway in the development of MDR is being studied extensively and some specific blockers of the pathway have been discovered. Some studies[9,10]suggest that modulation of JNK activation may be a new method to reverse MDR. However, how JNK activation is related to MDR, particularly in HCC cells, is still largely unknown. In this study, we provide the first direct evidence that JNK activity is negatively correlated with the degree of MDR in HCC cells.
Methods
Cell cultures
The human HCC cell line, SMMC-7721, was purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Science, the Chinese Academy of Sciences. Cells were cultured with RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented with 10% calf serum (HyClone) and maintained at 37 ℃ in a humidified atmosphere of 5% CO2/95% air. A multidrug-resistant human HCC cell line, SMMC-7721/ ADM, was developed by culturing SMMC-7721 cells in the presence of gradually increasing concentrations (0.01, 0.02, and 0.05 mg/L) of adriamycin (ADM, Zhejiang Hisun Pharmaceutical Co., Ltd., Zhejiang Province, China). The resistant cells were selected and resistance was maintained by culturing the cells in medium supplemented with 0.01, 0.02, or 0.05 mg/L adriamycin, and these MDR cell subclones were named SMMC-7721/ADM (0.01), SMMC-7721/ADM (0.02), and SMMC-7721/ADM (0.05).
Measurement of cellular sensitivity to anticancer drugs
The MTT [3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide] assay was used to determine drug sensitivity. SMMC-7721 and SMMC-7721/ADM (0.05) cells in the exponential growth phase were harvested by trypsinization (0.25% trypsin; HyClone) and seeded into 96-well plates at a concentration of 5× 103cells/200 μl/well. The cells were incubated overnight to allow attachment to the plates. On the next day, old medium was removed and fresh medium supplemented with various concentrations of anticancer drugs (adriamycin, fluorouracil, cisplatin, cyclophosphamide, mitomycin, or vincristine) was added to cultures and incubated for 24 hours. Control cells received fresh medium without anticancer drugs. After treatment, the medium was replaced with fresh drug-free medium (200 μl/well), and MTT (5 mg/ml in PBS; Sigma-Aldrich, St. Louis, MO, USA) was added to each well at a 1/10 dilution. After incubation for 4 hours at 37 ℃, the supernatant was carefully aspirated, 150 μl of DMSO was added to each well, and the plates were agitated to dissolve the crystal product. Absorbance was measured at 490 nm using a multi-well plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The IC50value, defined as the drug concentration required to reduce cell survival to 50% as determined by the relative absorbance of MTT, was assessed by probit regression analysis using SPSS 11.5 statistical software.
Flow cytometric analysis of cell cycle distribution
SMMC-7721 and SMMC-7721/ADM (0.05) cells were harvested, washed twice in ice-cold PBS, and fixed in 70% ethanol for 2 hours at 4 ℃. The cells were rehydrated with PBS and then incubated for 30 minutes at room temperature with propidium iodide staining solution [0.2 mg/ml propidium iodide, 0.2 mg/ml DNAse-free RNAse A (Roche, Basel, Switzerland) and 0.1% Triton X-100 in PBS]. By using red propidium-DNA fluorescence, 4000 events were acquired with an Epics XL flow cytometer (Beckman Coulter Inc., Fullerton, CA, USA) for each sample and the percentages of cells in the G0/G1, S, and G2/M phases of the cell cycle were determined by the system software (Beckman Coulter Inc.).
Flow cytometric analysis of P-gp and MRP1 expression levels
Flow cytometry was used to measure P-gp and MRP1 expression levels in HCC cells. The cultured SMMC-7721 and SMMC-7721/ADM (0.05) cells were collected by trypsinization, washed in ice-cold PBS, and then directly immunostained using FITC-conjugated anti-P-gp antibody (Cat. ab66250, Abcam plc, Cambridge, UK). Alternatively, the cells were first fixed and permeabilized with BDCytofix/CytopermTMsolution (Cat. No. 554722, BD Biosciences, San Jose, CA, USA) and then incubated with FITC-conjugated anti-MRP1 antibody (Cat. No. 557593, BD Biosciences). Flow cytometry was performed on the labeled cells as described above. An isotype control IgG (Cat. ab18455, Abcam plc) was evaluated in each experiment to determine the level of background fluorescence of negative cells. Mean fluorescence intensity was determined for positively stained cells.
RNA extraction and quantitative real-time polymerase chain reaction (PCR)
Cells were frozen in liquid nitrogen and stored at -80 ℃ to be used in real-time PCR experiments. JNK1, JNK2, and JNK3 mRNA expression levels were quantified by real-time PCR. Total cellular RNA was extracted using Trizol reagent (Gibco BRL, Life Technologies Inc., Rockville, MD, USA) according to the protocol recommended by the manufacturer. Briefly, the cells were homogenized with Trizol using a teflon homogenizer (Fisher Scientific, PA), then chloroform was added and mixed. The homogenate was centrifuged at 12 000 g for 10 minutes at 4 ℃ and the resulting aqueous phase was separated. The RNA was precipitated by mixing equal amounts of the aqueous phase and isopropanol. The sample was shaken, incubated for 20 minutes at room temperature, and centrifuged again as previously described. The pellet was washed with 75% ethanol, dissolved in DEPC-treated RNAse-free water, and stored at -70 ℃. The concentration of total RNA was determined by measuring the optical density at 260 nm using a Beckman Coulter DU 530 spectrophotometer.
Reverse transcription of total RNA to cDNA was carried out using a Gene Amp RNA PCR kit and a DNA Thermal cycler (Bio-Rad). Quantitative realtime PCR was performed with SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA), an ABI Prism 7700 real-time PCR machine (Applied Biosystems) and the following gene-specific primers: JNK1 GenBank: NM_002750 forward, 5'-GCA GGA ACG AGT TTT ATG ATG AC-3', and reverse, 5'-CTG GAA AGA GGA TTT TGT GGC-3'; JNK2 GenBank: NM_002752 forward, 5'-GCC AAC TGT GAG GAA TTA TGT CG-3', and reverse, 5'-GGT GGT GGG GCT TCT GCT T-3'; JNK3 GenBank: NM_002753 forward, 5'-ATC CTG GGG ATG GGC TAC A-3', and reverse, 5'-GAA GAG TTT GGG GAA GGT GAG T-3'; β-actin GenBank: NM_001101 forward, 5'-TGA CGT GGA CAT CCG CAA AG-3', and reverse, 5'-CTG GAA GGT GGA CAG CGA GG-3'. Results were analyzed with Sequence Detector software (v1.9, Applied Biosystems) and the specificity of amplified products was verified using Dissociation Curve Analysis software (v1.0, Applied Biosystems). The threshold for positivity of real-time PCR was determined based on negative controls. The mean Ct values from duplicate measurements were used to calculate the expression of the target gene with normalization to a housekeeping gene used as internal control (β-actin), and using the 2-ΔCtformula.
Western blotting analysis
The cultured SMMC-7721, SMMC-7721/ADM (0.01), SMMC-7721/ADM (0.02), and SMMC-7721/ADM (0.05) cells were lysed in radio-immuno-precipitation assay buffer [150 mmol/L NaCl, 50 mmol/L Tris (pH 8.0), 0.5% deoxycholic acid, 1% NP40 with 1 mmol/L phenylmethylsulfonyl fluoride]. By using a tissue lyser (Retsch GmbH, Haan, Germany) the samples were homogenized for 3 minutes at maximum frequency and then incubated for 2 hours at 4 ℃ with gentle rocking. Samples were then centrifuged at 12 000 g for 20 minutes and protein concentrations were measured in the supernatants (Protein Assay Dye, Bio-Rad). Cell extracts were denatured in LDS sample buffer for 5 minutes at 95 ℃, and subjected to SDS-PAGE (NUPAGE, 4%-12% Bis-Tris, Invitrogen, Carlsbad, CA, USA) and blotted onto PVDF membranes (0.2 μm, Invitrogen). Membranes were blocked with 5% milk in TBS-T (TBS containing 0.05% Tween 20) for 1 hour at room temperature and were subsequently incubated overnight at 4 ℃ with antibodies against JNK1 (Abcam plc.), JNK2, JNK3, or phospho-SAPK/ JNK (Thr183/Tyr185) (Cell Signaling Technology, Inc., Danvers, MA, USA). After incubation with the respective primary antibodies, membranes were incubated with species-specific horseradish peroxidase-labeled secondary antibodies. The membrane was developed using the ECL Plus Western blotting reagent (GE Healthcare, Little Chalfont, UK) and Fuji Film LAS-1000 equipment (Fuji Film, Tokyo, Japan). Similarly processed membranes were also incubated with 1∶5000 rabbit monoclonal antibodies to GAPDH (Cell Signaling Technology, Inc.) and HRP-coupled rabbit anti-mouse secondary antibodies to serve as controls. Calculations and statistical analysis were performed using ImageJ 1.37 software.
Statistical analysis
The results were expressed as mean±SEM. Values were analyzed using the statistical package SPSS for Windows Ver. 11.5 (SPSS Inc., Chicago, IL, USA). Statistical analyses were performed using Student'sttest when comparing two groups and ANOVA with Dunnett's post-test when comparing three or more groups.P<0.05 was considered statistically significant.
Results
Development of multidrug-resistant SMMC-7721/ADM subclones
MDR in HCC cells is a major obstacle in chemotherapy, however the mechanism of such resistance is still not completely understood. To study the regulation of MDR-related genes in HCC cells we first developed MDR in an HCC cell line (SMMC-7721) by treating the cells with gradually increasing concentrations of adriamycin. The morphological changes in cultured cells were continuously photographed during the period of cell proliferation from SMMC-7721 to SMMC-7721/ ADM (0.01) cells. We observed that the cells with lower drug resistance began to die 24 to 48 hours after the higher concentrations of adriamycin were added to the medium, and the mortality rate reached a peak about 72 hours later. To recover the proliferation ability, the remaining adherent cells needed to be cultured for at least 7 to 8 weeks (Fig. 1).
Determination of MDR
After culturing with adriamycin, cells were tested for their resistance to other anticancer drugs using a cytotoxicity assay. We found that besides adriamycin, SMMC-7721/ADM (0.05) cells were resistant to the anticancer drugs cisplatin, mitomycin, vincristine, cyclophosphamide, and fluorouracil. The lethal doses (IC50) of these drugs in SMMC-7721/ADM (0.05) cells were significantly higher than those in non-resistant SMMC-7721 cells (Table and Fig. 2).
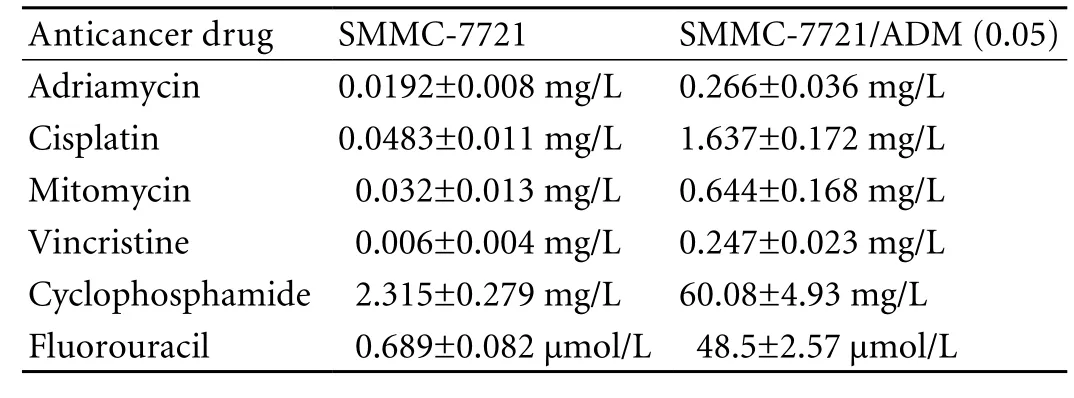
Table. Determination of IC50of anticancer drugs (n=5)
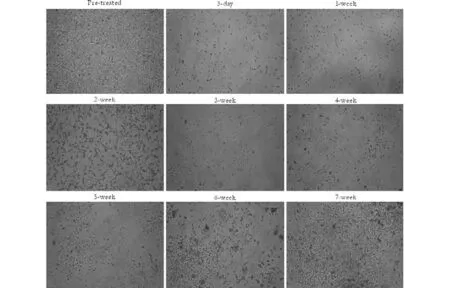
Fig. 1. Continuous photographic record of cell proliferation during the development of SMMC-7721/ADM (0.01). Adriamycin was added to SMMC-7721 cells at 0.01 mg/L and resistant cells were selected by removing the dead cells. The cells were continuously photographed during their progress from the parental SMMC-7721 to the resistant SMMC-7721/ADM (0.01) subclone. The resistant cells were cultured for about 7 weeks to confirm a stable phenotype.
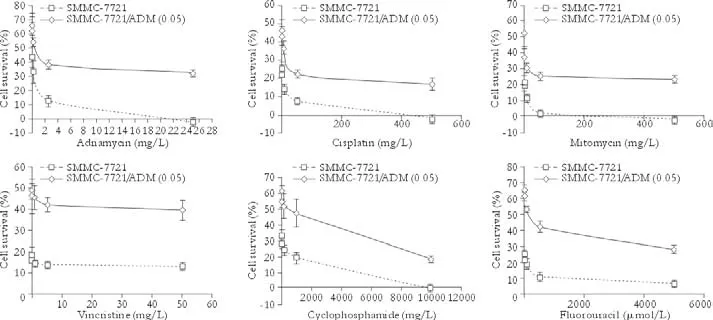
Fig. 2. Measurement of the sensitivity of SMMC-7721 and SMMC-7721/ADM (0.05) cells to anticancer drugs. Error bars (SEM) on the dose-response curves were derived from five independent experiments.
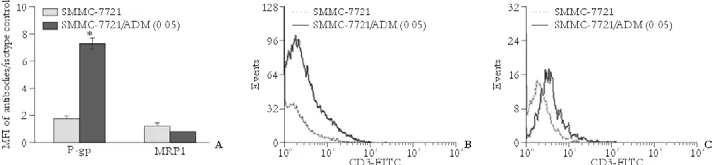
Fig. 3. P-gp and MRP1 expression in SMMC-7721 and SMMC-7721/ADM (0.05) cells. Mean fluorescence intensity is plotted against the Y-axis to depict the difference in expression of the two proteins in different cells (A) while the corresponding flow histograms are presented in the lower panels, P-gp (B) and MRP1 (C). *:P<0.001.
Cell cycle distribution
We investigated whether the cell cycle distribution was related to MDR. Previous studies[11,12]on the cell cycle distribution of cultured MDR cells reported diverse results. In this study, cell cycle phase distribution was assessed, using flow cytometry, to determine if MDR SMMC-7721/ADM (0.05) cells have cycle kinetics different from SMMC-7721 cells. An increase in the number of cells in the S phase was apparent (23.6± 0.93% vs. 17.86±1.91%,n=5,P<0.01). This effect was accompanied by a decrease in the percentage of cells in the G0/G1 phase (67.8±2.15% vs. 71.12±1.38%,n=5,P<0.05).
P-gp and MRP1 expression
To understand the mechanisms involved in the development of the acquired MDR of SMMC-7721/ ADM (0.05) cells, the expression levels of MDR-related proteins (P-gp and MRP1) were analyzed using flow cytometry. P-gp expression in MDR SMMC-7721/ADM (0.05) cells was over 4-fold higher than that in parental SMMC-7721 cells. There was no significant difference in MRP1 expression between these two cell lines (Fig. 3).
JNK1, JNK2, and JNK3 mRNA expression in parental and MDR cells
To determine the role of JNK signaling in the development of MDR, we assessed JNK1, JNK2, and JNK3 mRNA expression in parental and resistant cells (Fig. 4). Quantitative real-time PCR analysis demonstrated that JNK1, JNK2, and JNK3 mRNA expression in all resistant cells was lower than that in theparental cell line, except for one subclone, SMMC-7721/ ADM (0.02) (Fig. 4).
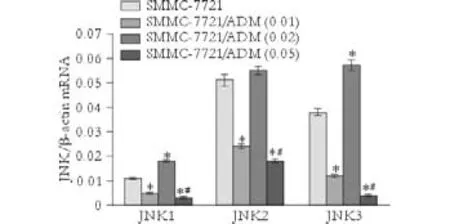
Fig. 4. JNK1, JNK2, and JNK3 mRNA expression in parental and MDR cells. Target gene expression in each sample was normalized to β-actin mRNA expression in the same sample. *:P<0.001, compared with SMMC-7721; #:P<0.01, compared with SMMC-7721/ADM (0.01).
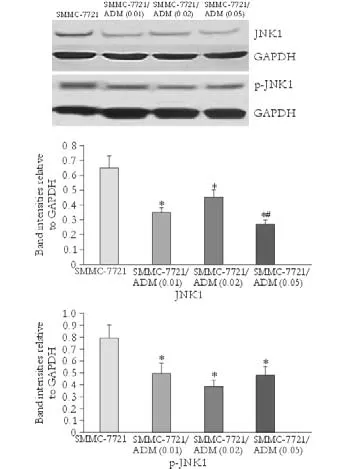
Fig. 5. Expression and phosphorylation of JNK1 in parental and MDR cells. Levels of total and phosphorylated JNK1. The upper panel shows protein bands developed by Western blotting while the lower two panels show calculated band intensity for quantitative comparison. *:P<0.05, compared with SMMC-7721; #:P<0.05, compared with SMMC-7721/ADM (0.01).
Expression and phosphorylation of JNK1, JNK2, and JNK3 in parental and MDR cells
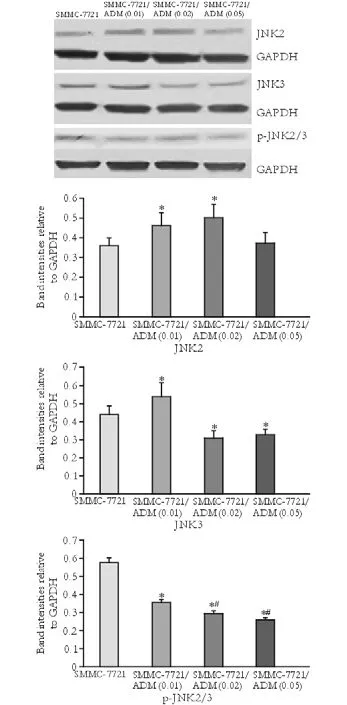
Fig. 6. Expression and phosphorylation of JNK2, JNK3 in parental and MDR cells. Protein bands for JNK2 and JNK3 and their phosphorylated forms (upper panel) and corresponding charts from calculated band intensities (lower panels). *:P<0.05, compared with SMMC-7721; #:P<0.05, compared with SMMC-7721/ADM (0.01).
Western blotting analysis of JNK1, JNK2, JNK3, and p-JNK was performed in SMMC-7721, SMMC-7721/ ADM (0.01), SMMC-7721/ADM (0.02) and SMMC-7721/ADM (0.05) MDR cells. Compared to the parental cell line, expression and phosphorylation of JNK1 were markedly lower in resistant cells. However, the extent of this difference did not correlate with the concentration of the drug, to which the cells were resistant (Fig. 5). In general, JNK3 expression was decreased in MDR cells while JNK2 expression was increased. Compared to the control group, reduced phosphorylation of JNK2/3 was detected in cells with MDR (Fig. 6).
Discussion
Previously, we have shown that ERK5 is involved in MDR, and ERK1/2 activity is down-regulated in HCC cells with P-gp-mediated MDR.[13,14]Besides ERK, other kinases such as JNK have been implicated in hypoxiainduced drug resistance.[15,16]JNK is a member of the mitogen-activated protein kinase family that binds the NH2-terminal activation domain of the transcription factor c-Jun and phosphorylates c-Jun.[17]The activity of JNK has been implicated in the regulation of embryonic morphogenesis, cell proliferation, tumor transformation, and apoptosis. A study[18]has shown that cancer cells overexpressing P-gp have low levels of endogenous JNK activity, and that c-Jun plays a critical role in the downregulation of P-gp by salvicine, a synthetic diterpenoid quinine. Although the major body of evidence argues that JNK is implicated in MDR, studies[19,20]indirectly suggest that c-Jun activation is negatively correlated with human MDR1 gene expression. It is unclear at present whether the discrepancy between these studies is due to the fact that JNK differentially regulates MDR1 gene expression under different conditions or merely because of the variations in experimental protocols, such as the use of different cell lines. To bridge this gap in our current knowledge about the relationship between MDR and JNK, we studied the expression and phosphorylation of the crucial kinases of the JNK pathway, JNK1, JNK2, and JNK3 in HCC cell lines with different degrees of MDR. We also attempted to identify the specific molecular aspects that govern this correlation in HCC cell lines. Understanding the involvement of the JNK pathway in MDR might be useful in future therapy for HCC.
Adriamycin is a chemotherapeutic agent principally used for the treatment of solid tumors including HCC.[21]Although it can work through various mechanisms, resistance to adriamycin still develops in a broad range of cell lines.[22,23]We established several drug-resistant HCC cell subclones by culturing the cells in the presence of increasing concentrations of adriamycin. The IC50of anticancer drugs to the MDR SMMC-7721/ADM (0.05) subclone was much higher than that to parental SMMC-7721, i.e. 19.14-fold for adriamycin, 33.89-fold for cisplatin, 20.13-fold for mitomycin, 41.16-fold for vincristine, 25.95-fold for cyclophosphamide, and 70.39-fold for fluorouracil. This finding showed that the acquired MDR of the SMMC-7721/ADM (0.05) subclone was stable.
We found a clear difference in the cell cycle distribution between MDR SMMC-7721/ADM (0.05) cells and the parental cell line. A significant increase in the percentage of cells in the S phase was accompanied by a decrease in the percentage in the G0/G1 phase of resistant SMMC-7721/ADM (0.05) cells that probably contributed to the lower proliferation ability of these cells. Moreover, a delay in the cell cycle may lead to cellular escape from the cytotoxicity of cell cyclespecific agents (e.g. vincristine and fluorouracil) and the development of MDR.[24,25]Both P-gp and MRP1 are ATP-binding cassette transporter proteins. Over-expression of such proteins is one of the major mechanisms that contribute to the MDR phenotype. Both P-gp and MRP1 function as drug efflux pumps that actively transport drugs from the inside of cells to the outside and interfere with the intracellular accumulation of drugs necessary for cancer cell killing. Our results showed that expression of P-gp in SMMC-7721/ADM (0.05) cells was over 4-fold higher than that in parental SMMC-7721 cells, but the MRP1 expression was similar, which suggests that overexpression of P-gp but not MRP1 mainly contributes to the MDR of SMMC-7721/ADM (0.05) cells. This finding is consistent with the fact that high P-gp expression is seen along with low MRP1 expression in liver tissue as well as in HCC cell lines.[26]
Three closely related JNK genes (1, 2 and 3) give rise to different splice variants resulting in ten potential isoforms.[27]The physiological reasons for the existence of so many isoforms are not known and the nature of the JNK proteins expressed by different tissues remains unclear. At present, it is well accepted that JNK1, JNK2, and JNK3 are isozymes and all are expressed in cultured cell lines. While JNK1 and JNK2 are extensively expressed in mammalian tissues, significant expression of JNK3 is restricted to certain tissues such as the brain and testis.[28-30]To investigate whether or not all JNKs are involved in the P-gp-mediated MDR in HCC cells, we compared the extent of mRNA expression, protein expression, and phosphorylation of JNK1, JNK2, and JNK3 between parental and resistant cell lines. The results showed that JNK1, JNK2, and JNK3 mRNA expressions were reduced as resistance developed in HCC cells. Protein expression was decreased forJNK1 and JNK3 and increased for JNK2 in MDR cells, compared to expression in the parental cell line. However, the extent of changes in protein expression was not correlated with the degree of drug resistance of the cell lines. The extent of phosphorylation of JNK1, JNK2, and JNK3 was decreased in SMMC-7721/ADM MDR cells. Moreover, HCC cells with a higher degree of resistance showed less phosphorylation of JNK2/3. This evidence suggests that JNK1, JNK2, and JNK3 activities are negatively correlated with the degree of MDR in HCC cells.
In this study, we successfully established SMMC-7721/ADM (0.01), SMMC-7721/ADM (0.02) and SMMC-7721/ADM (0.05) multidrug-resistant HCC cell subclones. We demonstrated that the MDR of SMMC-7721/ADM (0.05) cells is associated with the overexpression of P-gp but not MRP1, and that JNK1, JNK2, and JNK3 activities are negatively correlated with the degree of MDR in HCC cells. These findings provide new insights into the complicated regulatory mechanisms of the MDR phenotype and suggest that JNK1, JNK2, and JNK3 are potential drug targets for circumventing MDR in HCC.
Acknowledgement
We appreciate helpful discussion with Dr. Anirban Roy from the Center for Neurovirology, Thomas Jefferson University, Philadelphia, Pennsylvania, USA.
Funding: This study was supported by grants from the Medical Innovation Fundation of Fujian Province (No. 2007-CXB-7), and the Natural Science Foundation of Fujian Province (No. 2009D010). Ethical approval: Not needed.
Contributors: WXM proposed the study. YF wrote the first draft. YF and MQM analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. YF is the guarantor.
Competing interest: No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology 2002;36:S74-83.
2 But DY, Lai CL, Yuen MF. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol 2008;14: 1652-1656.
3 El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 2003;139:817-823.
4 Wakamatsu T, Nakahashi Y, Hachimine D, Seki T, Okazaki K. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins. Int J Oncol 2007;31:1465-1472.
5 Pérez-Tomás R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem 2006;13:1859-1876.
6 Angelini A, Iezzi M, Di Febbo C, Di Ilio C, Cuccurullo F, Porreca E. Reversal of P-glycoprotein-mediated multidrug resistance in human sarcoma MES-SA/Dx-5 cells by nonsteroidal anti-inflammatory drugs. Oncol Rep 2008;20: 731-735.
7 Yan F, Jiang Y, Li YM, Zhen X, Cen J, Fang WR. Reversal of P-glycoprotein and multidrug resistance-associated protein 1 mediated multidrug resistance in cancer cells by HZ08 Isomers, tetrataisohydroquinolin derivatives. Biol Pharm Bull 2008;31:1258-1264.
8 Stierlé V, Duca M, Halby L, Senamaud-Beaufort C, Capobianco ML, Laigle A, et al. Targeting MDR1 gene: synthesis and cellular study of modified daunomycin-triplexforming oligonucleotide conjugates able to inhibit gene expression in resistant cell lines. Mol Pharmacol 2008;73: 1568-1577.
9 Hu XF, Li J, Yang E, Vandervalk S, Xing PX. Anti-Cripto Mab inhibit tumour growth and overcome MDR in a human leukaemia MDR cell line by inhibition of Akt and activation of JNK/SAPK and bad death pathways. Br J Cancer 2007;96: 918-927.
10 Zhou J, Liu M, Aneja R, Chandra R, Lage H, Joshi HC. Reversal of P-glycoprotein-mediated multidrug resistance in cancer cells by the c-Jun NH2-terminal kinase. Cancer Res 2006;66:445-452.
11 Lukyanova NY, Rusetskya NV, Tregubova NA, Chekhun VF. Molecular profile and cell cycle in MCF-7 cells resistant to cisplatin and doxorubicin. Exp Oncol 2009;31:87-91.
12 Li Y, Li S, Han Y, Liu J, Zhang J, Li F, et al. Calebin-A induces apoptosis and modulates MAPK family activity in drug resistant human gastric cancer cells. Eur J Pharmacol 2008;591:252-258.
13 Yan F, Wang XM, Pan C, Ma QM. Down-regulation of extracellular signal-regulated kinase 1/2 activity in P-glycoprotein-mediated multidrug resistant hepatocellular carcinoma cells. World J Gastroenterol 2009;15:1443-1451.
14 Yan F, Wang XM, Pan C. Expression of ERK5 in multidrugresistant hepatocellular carcinoma cell line. Nan Fang Yi Ke Da Xue Xue Bao 2009;29:483-486.
15 Comerford KM, Cummins EP, Taylor CT. c-Jun NH2-terminal kinase activation contributes to hypoxia-inducible factor 1alpha-dependent P-glycoprotein expression in hypoxia. Cancer Res 2004;64:9057-9061.
16 Liu M, Li D, Aneja R, Joshi HC, Xie S, Zhang C, et al. PO(2)-dependent differential regulation of multidrug resistance 1 gene expression by the c-Jun NH2-terminal kinase pathway. J Biol Chem 2007;282:17581-17586.
17 Maruyama J, Naguro I, Takeda K, Ichijo H. Stress-activated MAP kinase cascades in cellular senescence. Curr Med Chem 2009;16:1229-1235.
18 Miao ZH, Ding J. Transcription factor c-Jun activation represses mdr-1 gene expression. Cancer Res 2003;63:4527-4532.
19 Kang CD, Ahn BK, Jeong CS, Kim KW, Lee HJ, Yoo SD, etal. Downregulation of JNK/SAPK activity is associated with the cross-resistance to P-glycoprotein-unrelated drugs in multidrug-resistant FM3A/M cells overexpressing P-glycoprotein. Exp Cell Res 2000;256:300-307.
20 Wartenberg M, Ling FC, Schallenberg M, Baumer AT, Petrat K, Hescheler J, et al. Down-regulation of intrinsic P-glycoprotein expression in multicellular prostate tumor spheroids by reactive oxygen species. J Biol Chem 2001;276: 17420-17428.
21 Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004;56:185-229.
22 Barrand MA, Heppell-Parton AC, Wright KA, Rabbitts PH, Twentyman PR. A 190-kilodalton protein overexpressed in non-P-glycoprotein-containing multidrug-resistant cells and its relationship to the MRP gene. J Natl Cancer Inst 1994;86: 110-117.
23 Mehta K. High levels of transglutaminase expression in doxorubicin-resistant human breast carcinoma cells. Int J Cancer 1994;58:400-406.
24 Rebbaa A. Targeting senescence pathways to reverse drug resistance in cancer. Cancer Lett 2005;219:1-13.
25 Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta 2007;1775:5-20.
26 Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105-127.
27 Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 1996;15:2760-2770.
28 Finch A, Davis W, Carter WG, Saklatvala J. Analysis of mitogen-activated protein kinase pathways used by interleukin 1 in tissues in vivo: activation of hepatic c-Jun N-terminal kinases 1 and 2, and mitogen-activated protein kinase kinases 4 and 7. Biochem J 2001;353:275-281.
29 Shibata W, Maeda S, Hikiba Y, Yanai A, Sakamoto K, Nakagawa H, et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res 2008;68:5031-5039.
30 Bjorkblom B, Vainio JC, Hongisto V, Herdegen T, Courtney MJ, Coffey ET. All JNKs can kill, but nuclear localization is critical for neuronal death. J Biol Chem 2008;283:19704-19713.
Received September 23, 2009
Accepted after revision March 3, 2010
Author Affiliations: Department of Gastrointestinal Surgery (Yan F, Liu ZC and Yuan SB), Department of Hepatobiliary Surgery (Wang XM and Ma QM), and Department of Pathology (Pan C), Zhongshan Hospital, Xiamen University, Xiamen 361004, China; Xiamen University Digestive Diseases Institute; Digestive Diseases Center of Xiamen City, Xiamen 361004, China (Yan F, Wang XM, Liu ZC and Yuan SB)
Feng Yan, MD, PhD, Department of Gastrointestinal Surgery, Zhongshan Hospital, Xiamen University, Xiamen 361004, China (Tel: 86-592-2292517; Fax: 86-592-2212328; Email: yanfeng@xmzsh.com)
© 2010, Hepatobiliary Pancreat Dis Int. All rights reserved.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Gallbladder cancer with tumor thrombus in the superior vena cava
- Budd-Chiari syndrome secondary to caval recurrence of renal cell carcinoma
- A prospective study on radiofrequency ablation locally advanced pancreatic cancer
- Liver graft vascular variant with 3 extra-hepatic arteries
- An effective model for predicting acute kidney injury after liver transplantation
- Pancreas transplantation in the mouse