盐酸左氧氟沙星含量测定方法研究
2010-06-01许晓文
许晓文,袁 林
(1.广东省佛山市药品检验所,广东 佛山 528000; 2.中澳合资东方澳龙制药有限公司,广东 佛山 528234)
2000版《中国药典》(2004年增补本)采用高效液相色谱(HPLC)法测定盐酸左氧氟沙星制剂的含量[1],但该法使盐酸左氧氟沙星及其主要杂质的保留时间过长,工作效率影响极大,消耗增加流动相。笔者研究认为,适当提高流动相中甲醇所占的比例,系统适用性可满足药典要求,样品的分析时间可缩短60%以上,含量测定结果与中国药典方法无明显差异,能更好地适用于常规检验工作和厂方大批量生产时的质量控制。现报道如下。
1 仪器与试药
Waters 510型高效液相色谱仪(SEPU3000工作站,WatersTM486 Tunable Absorbance Detector);Waters Temperature Control Module;Mettler AE 240型电子天平。左氧氟沙星对照品(中国药品生物制品检定所,批号为130455-2006049,含量为97.3%);盐酸左氧氟沙星制剂(市场抽样);己烷磺酸钠(分析纯,山东禹王实业有限公司化工分公司);磷酸二氢钾(分析纯,广州光华化学厂有限公司);磷酸(分析纯,广州化学试剂厂);甲醇(色谱纯,天津市四友精细化学品有限公司);水为超纯水;0.45 μm微孔滤膜。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:大连依利特Kromasil ODS2 柱(200 mm×4.6 mm,5 μm);流动相:己烷磺酸钠溶液[取己烷磺酸钠0.98 g,加磷酸盐缓冲液(取磷酸二氢钾6.8 g,加水溶解并稀释至1 000 mL,加0.05 mol/L磷酸约 500 mL,调节 pH 至 2.4)1 000 mL,使溶解,摇匀]-甲醇(2 ∶2);检测波长:293 nm;流速:1.0 mL/min;柱温:40℃。取左氧氟沙星对照品的水溶液(1→1 000)10 mL,置试管中,用日光灯(3 500 lx)照射3 h,取该溶液10 μL注入液相色谱议,记录色谱图,结果相对于主峰保留时间约为1.2倍处检测到杂质峰;另取左氧氟沙星对照品溶液连续进样4次,其 RSD为0.8%;理论塔板数按左氧氟沙星峰计算高于1 500。
2.2 溶液制备
取左氧氟沙星对照品约10 mg,精密称定,置20 mL量瓶中,加0.03 mol/L盐酸溶液约15 mL,置超声波浴中使溶解,再加0.03 mol/L盐酸溶液稀释至刻度,摇匀,滤过,精密量取续滤液10 mL置25 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,作为对照品溶液。取相当于左氧氟沙星50 mg的样品,精密称定,置50 mL量瓶中,加0.03 mol/L盐酸溶液约35 mL,置超声波浴中使溶解,再加0.03 mol/L盐酸溶液稀释至刻度,摇匀,滤过,精密量取续滤液10 mL置50 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,作为供试品溶液。
2.3 本方法与药典方法比较
2.3.1 色谱图
图1和图2分别是采用本方法和药典方法测定左氧氟沙星对照品的色谱图。可见,左氧氟沙星在两图中均呈现良好峰形,且与杂质分离效果良好,但保留时间有很大差异,前者为8.63 min(图1 A),后者为21.06 min(图2),较后者缩短了约60%的时间。说明在甲醇比例提高后,对杂质的分离能力无显著影响,但保留时间有很大地缩短。图1 B是采用本方法确定左氧氟沙星样品的色谱图,可见左氧氟沙星峰形尖锐,与杂质分离良好。说明该方法适合于左氧氟沙星样品的测定。
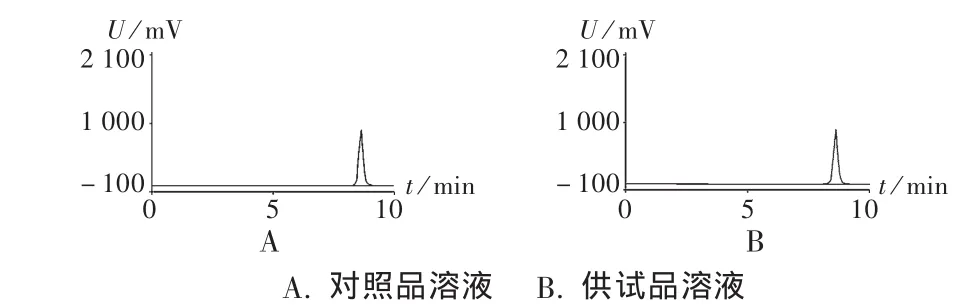
图1 高效液相色谱图(本方法)
2.3.2 含量测定结果
分别采用药典方法和本方法测定了几批盐酸左氧氟沙星制剂中左氧氟沙星的含量,结果药典方法为99.5% ,97.2% ,98.3%,95.6%;本方法为99.8%,97.0%,97.9%,95.8%。应用 F检验,结果 F=1.03,对于 α =0.05,查表得 F1-0.05/2(3,3)=9.28,F < F1-0.05/2(3,3),说明两种方法的精密度无显著差异。应用 t检验,结果 t=0.025,查表得 t1-0.05/2(6)=2.447,t值远小于 t1-0.05/2(6),说明两种方法的平均值之间也无显著差异。
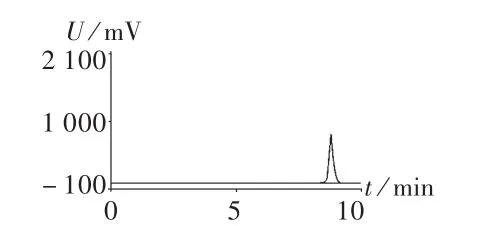
图2 高效液相色谱图(药典法)
3 讨论
比较理想的流动相应当具有稳定性好、廉价易得、对环境无污染、在检测波长区域无吸收、分离效果好、样品组分保留时间短、分析速度快等特点[2]。中国药典所规定的流动相体系不失为一种较理想的流动相体系,但左氧氟沙星的保留时间较长,这一大缺陷。左氧氟沙星的保留时间随流动相中甲醇比例的增大而缩短。将己烷磺酸钠溶液-甲醇流动相的比例由3∶1调整为2∶2,使甲醇的含量由原来的33%提高到50%,可在保证系统适用性符合中国药典规定的前提下,使左氧氟沙星的保留时间缩短了约60%。
与中国药典方法比较,本方法一方面继承了原方法的优点,另一方面改进了原方法的一些缺陷,在保证分离效果的前提下,缩短了组分的保留时间,提高了分析速度,含量测定结果与中国药典无显著差异。因此,本方法能更好地适用于常规检验工作和厂方大批量生产时的质量控制。
[1]国家药典委员会.中华人民共和国药典(二部)2000版2004年增补版[M].北京:化学工业出版社,2004:57.
[2]郭 磊,张凌艳.头孢拉定含量测定方法的研究[J].黑龙江医药,2003,16(5):424-425.
