基于肠道菌群多模态信息融合的疾病检测方法
2024-07-05刘畅吴舜尧
刘畅 吴舜尧

摘要:利用可操作分类单元(Operational Taxonomical Units,OTU)或扩增子序列变体(Amplicon Sequence Variants,ASV)推断下游信息时,现有扩增子测序数据分析可能丢失不同物种谱构建法的多模态信息,为此,详细分析了4种疾病的OTU和ASV数据集在肠道群落多样性和群落结构方面的差异,提出了一种有效整合OTU与ASV用于疾病表征预测的方法:MDDMI(Microbiome-based Disease Detection with Multimodal Information)。实验结果表明,MDDMI优于单模态数据分析法。
关键词:图卷积神经网络;疾病预测;多模态;肠道菌群
中图分类号:TP183
文献标志码:A
肠道菌群是人体内最大的微生物群落,与人体健康息息相关[1],同时与多种疾病亦有紧密关联,例如炎症性肠病、肥胖、糖尿病、心血管疾病、神经系统疾病等。研究肠道菌群变化能够预测相关疾病的发生,改善治疗效果。利用微生物组学数据检测疾病已成为生物医学领域的研究热门,例如将深度学习应用于人类肠道微生物组丰度表进行疾病预测[2-3];利用自助法对16S rRNA序列进行子采样,计算子采样序列的k-mer并利用其多态性预测疾病[4];将微生物系统发育树和微生物分类群的相对丰度表示在单个矩阵中,使用卷积神经网络(Convolutional Neural Networks,CNN)解决疾病预测问题[5]。这些预测方法只使用了单一模态数据,并未有效利用不同测序分析方法的多模态信息。从使用可操作分类单元(Operational Taxonomic Units,OTU)法转为使用扩增子序列变体(Amplicon Sequence Variants,ASV)法进行微生物组学分析是近年来的发展趋势[6-7]。OTU分析方法是将序列按照一个相似性阈值(通常为97%)使用匹配算法进行聚类[8-9],无法考虑到较小生物学变异信息。最近相关研究通过ASV法克服这一缺点,与OTU聚类方法不同,ASV是基于序列的变异体(即序列的不同形式)进行聚类,在一定程度上减少了假阳性序列[10]。OTU或ASV法得出的生物学解释和结论存在差异[11-15],因此考虑将两种方法得到的数据有效整合。已有研究融合肠道微生物多模态数据预测疾病,例如使用变分自编码器融合宏基因组测序得到肠道微生物物种丰度和菌株标记物,在6种疾病的5个队列中,AUC值比使用单一模态数据高[16]。本文提出了一种使用图卷积神经网络(Graph Convolutional Networks,GCN)融合OTU与ASV的方法,分别对美国肠道计划(American Gut Project,AGP)[17]的4种疾病数据集的多模态数据进行融合验证,并使用不同方法对4种疾病数据集得到的群落多样性以及属水平菌群结构差异进行分析。
1 AGP数据集中OTU与ASV法的差异
AGP数据集中共有21种疾病,选取其中患病样本量较多且较为常见的4种疾病(Autoimmune、Cancer、Lung Disease、IBS)进行分析。
1.1 Autoimmune数据集中OTU与ASV法的差异
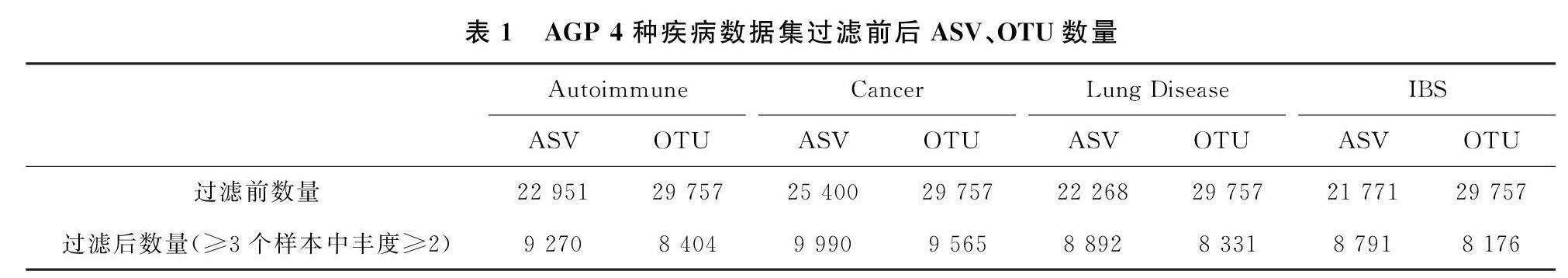
4种疾病中Autoimmune患病样本量最多,且患病率最高,对比使用OTU与ASV法对该疾病得到的群落多样性以及属水平菌群结构差异。初步过滤后(去除所有样本中都没出现过的OTU和ASV),ASV数量比OTU少。随后应用技术过滤去除伪序列(序列至少在3个独立样本中出现且每个样本中至少有2个计数),ASV减少数量低于OTU,表明低丰度OTU占比较大(表1)。
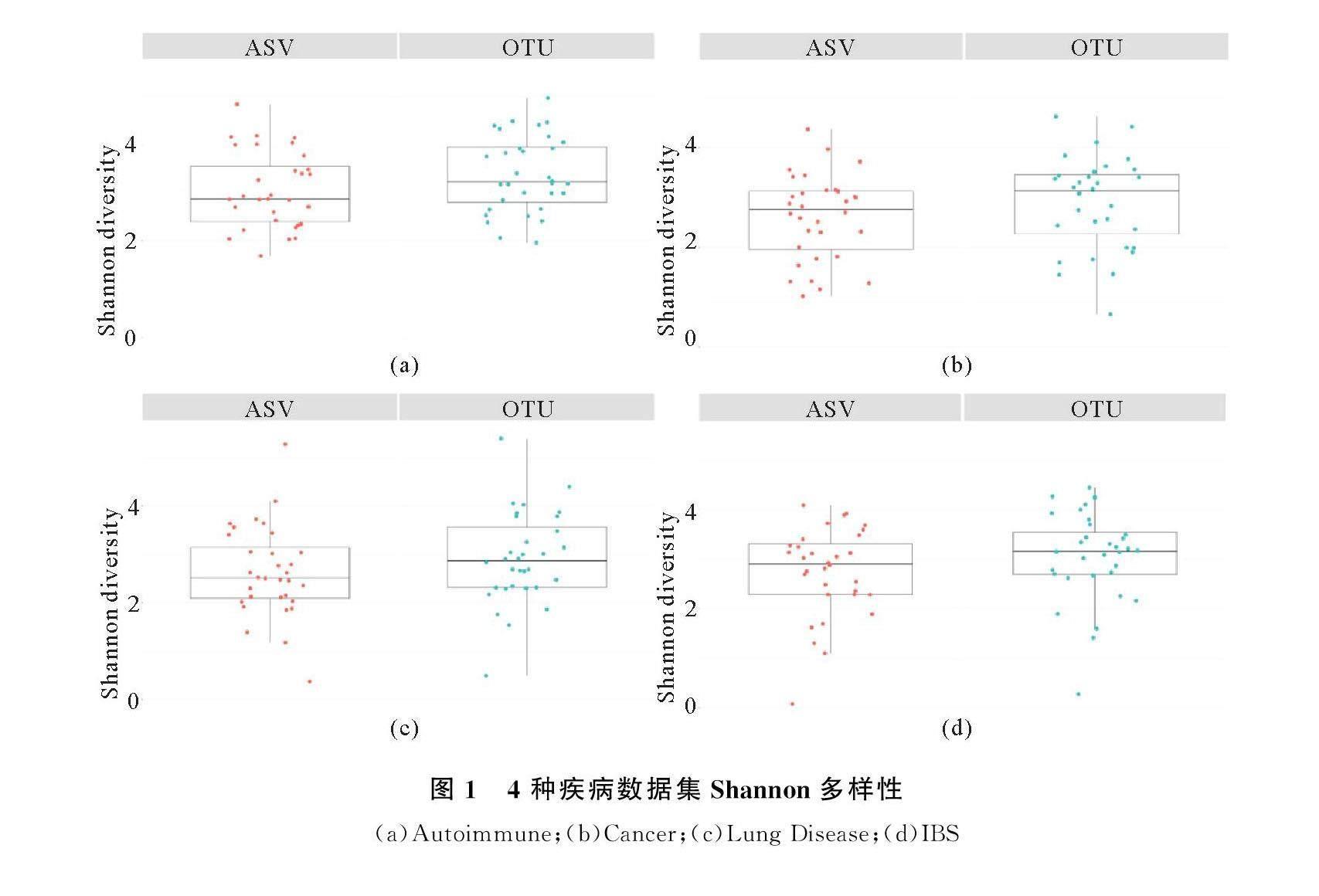
随机选取16个样本对OTU与ASV数据集(下文记为o与a数据集)进行群落多样性分析,a数据集的Shannon指数[18]低于o数据集,表明OTU法精确度有待进一步探究(图1(a))。在测序深度达到15 000个序列后,a数据集的物种丰富度达到平台期,而o数据集的物种丰富度继续增加(图2(a))。基于相同的测序深度, ASV法比OTU法得到的曲线收敛更快,表明ASV法在识别微生物群落时具有更高的分辨率和准确性,能够高效识别原始测序数据中微生物的物种或亚物种。
使用OTU与ASV法分析Autoimmune数据集属水平菌群,ASV法共获得716个属,OTU法共获得893个属,有670个重叠属(图3(a))。其中只存在于OTU法的223个属丰度均较低,细菌群落中RA(Rel. Abundance)均小于0.5%,且RA小于0.1%的占多数,只存在于ASV法得到的属的情况与之相似,在细菌群落中RA均较低。
对o与a数据集分别进行显著性分析,ASV法得到的显著属数量较多(ASV:29,OTU:8),其中只有两个与OTU法得到的显著属重叠,且OTU法得到的显著属大多丰度较低(RA<0.1%)。
由于两种方法的差异主要来源于低丰度属,对RA高于0.1%的属进行重分析。应用0.1%阈值过滤后,OTU与ASV法分别得到4个和3个显著属,其中没有显著属重叠。表明OTU与ASV法得到的生物学结论确实存在差异,与两者原理本质不同有关。
1.2 其余3种数据集中OTU与ASV法的差异
为验证Autoimmune数据集中得到的结论,使用OTU与ASV法对其余3种疾病得到的群落多样性以及属水平菌群结构差异进行分析。同Autoimmune数据集分析方法,初步过滤后,ASV数量均少于OTU。过滤伪序列后,ASV数量也均少于OTU(表1)。3种疾病的a数据集Shannon指数和丰富度均低于o数据集(图1),丰度曲线收敛均快于o数据集(图2)。3种疾病数据集中,使用OTU法检测到的属数量均多于ASV法,其中只存在于ASV法得到的属与只存在于OTU法得到的属RA均较低。OTU与ASV法对高丰度属的检测能力相近,例如对Cancer数据集进行过滤(RA<0.1%),使用OTU法得到79个属,使用ASV法得到74个属,其中66个重叠(图3)。可知,不同方法产生的差异主要源于RA水平较低(<0.1%)的属。
OTU或ASV法会得到不同的微生物多样性、丰富度、组成结构和差异丰度,从而影响生物学结论。显著性分析时,相较于OTU法,过滤RA低于0.1%和0.5%的属后,ASV法得到的显著属数量仍较少。因此,对OTU法得到的数据集进行分析时建议严格过滤。两种方法的差异主要源于低丰度属,因此分析低丰度属或种时需谨慎。AGP的4种疾病数据集中,a数据集的多样性和物种丰富度均低于o数据集,这与高比例(约39.7%±6.1%)的ASV未能被准确注释有关,这些未分配的ASV可能为新微生物变异体,或是数据库中尚未记录的微生物序列,而OTU法在全部序列聚类时,纳入了数据库中未包含的非生物序列或与目标微生物无关的序列。同时,使用ASV法得到的数据集丰富度-测序深度曲线收敛比OTU法快,因为ASV法能更好地捕获物种信息,这也是a数据集α多样性较低的原因。
同一数据集使用OTU与ASV法会得到不同群落组成,产生不同生物学结论,ASV法可以更准确地识别和分类微观生物变异体,OTU法则可以更有效地识别和分类相似微生物种类,通过多模态融合,结合两种方法的优势,从而更准确地识别和分类样本中的微生物群落,提高疾病预测效果。
2 MDDMI模型
2.1 模型输入层
分别获取OTU与ASV法得到的微生物转移网络中的所有样本生物分类单元组成,使用parallel-meta[19]工具对OTU法得到的生物分类单元组成样本及丰度进行整合,生成包含所有样本OTU丰度信息的丰度矩阵。由于OTU与ASV丰度矩阵中存在大量丰度值为0的向量,直接用作节点特征可能影响预测结果,通过计算所有矩阵向量与节点标签之间的相关性,剔除相关性较低的向量,获取显著节点并降维初始特征。
2.2 模型框架层
构建OTU与ASV微生物转移网络系统发育树模型,记为G=(V,E),其中V表示节点,E表示节点之间的连边,代表节点之间的相关性。
MDDMI按照属信息构造OTU与ASV连边,使用GCN获取相关性。GCN是CNN的变形,可以解决CNN无法处理的非结构化数据。在GCN中,节点通过聚合其邻居节点特征信息更新自身节点特征信息,通过损失函数,反向传播更新所有网络中的可学习参数,并以此学习节点的嵌入向量表示。GCN的原理为
H(l+1)=σ(D-12A~D-12H(l)W(l)+bl)(1)
其中,H(l)为图内节点第l层的向量表示,D为图的度矩阵,A~为图的邻接矩阵,W(l)为可训练的线性变换权重参数,bl为偏置项,σ为非线性激活函数,例如ReLU,H(l+1)由H(l)计算。
将GCN学习到的图嵌入向量与显著属信息(累加所有出现在显著属列表的特征丰度)添加至o与a数据集中,使用深度神经网络(Deep Neural Networks,DNN)[2]进行疾病预测(图4)。
2.3 预测层
DNN输出的二维特征中第一维度特征值作为预测得分,使用binary_cross_entropy作为目标损失函数进行训练
Loss=-1N∑Ni=1yi×log(p(yi))+(1-yi)×log(1-p(yi))(2)
其中,y为正负样本标签0或1,代表样本是否患病,p(y)为输出属于y标签的概率。预测得分越高,节点标签为正样本的概率越大,即该样本为患病样本的可能性越大。
3 实验设置与结果讨论
3.1 数据获取与预处理
本文使用AGP调查问卷中29 346个样本的表型数据作为标准判断样本是否患病,并处理调查问卷中的微生物样本采集信息,使用Vsearch[20]获取其中26 970个样本的OTU组成信息,使用parallel-meta自动分析流程进行处理,生成包含26 970个样本在内的OTU相对丰度表;同时使用Deblur[21]获取样本的ASV组成信息。
Autoimmune、Cancer、Lung Disease和IBS 4种疾病数据集创建方式如下:从表型数据中分别得到“Autoimmune”、“Cancer”、“Lung Disease”和“IBS”列中“被专家确诊”的样本作为患病样本;在IBS、Autoimmune、Thyroid等二十余种疾病上都表现为健康表型的样本作为健康样本。将所有健康样本分别与4种疾病数据集的患病样本合并,得到4种疾病的初始样本。由于不同居住环境会导致人体内菌群产生较大差异,为避免居住地等因素对样本菌群组成产生影响,对4种疾病的样本分别进行预处理,只保留居住地为“USA”的样本以保证菌群组成整体相似性。筛选后的4种疾病样本中,只保留健康样本中1 500个作为最终健康样本,以避免数据集不平衡。根据得到的4种疾病样本编号从OTU、ASV组成信息中提取4种疾病数据集的o、a数据集。
3.2 实验参数与比较模型
将MDDMI与两种基础方法(RF(Random Forest)[2]、DNN)进行比较。RF处理高维数据时性能良好,处理非线性关系的能力也较强,能够自动处理特征选择和数据平衡问题,具有较好鲁棒性。DNN在各种任务中表现出色,能够学习多个层次的抽象特征表示,具有较强建模能力,可以自动学习输入数据中的复杂模式和关系,适用于处理大规模高维数据。
3.3 评估指标
评估指标反映模型对样本数据的处理效果,通过对比模型预测结果与真实标签,计算模型的预测准确率等指标,衡量模型解决问题的能力。接受者操作特征曲线是一种有效的二分类模型性能评估方法,通过设置阈值,将样本分为患病和健康两类,利用ROC曲线和AUC值衡量模型预测精度。采用五折交叉验证法,使用sklearn中的StratifiedKFold包保证数据均匀分布和结果准确性。通过比较各个模型的AUC值评估疾病检测模型性能表现。
3.4 结果对比与讨论
Autoimmune数据集运行结果表明,两种基础模型预测得到的AUC值相当。为验证物种注释覆盖率对使用a数据集训练效果的影响(仅有60%的ASV被正确物种注释),使用所有的ASV训练两种基础模型,发现物种注释覆盖率对预测结果影响较小。此外,使用o数据集训练基础模型,预测得到的AUC值高于a数据集,使用MDDMI得到的AUC值高于使用任一单一模态数据集,提升至0.768(图5(a))。上述AUC值为五次五折交叉验证结果的均值。
为验证模型泛化能力,继续对AGP数据集的Cancer、Lung Disease和IBS进行预测(图5(b)-(d))。仅在Lung Disease预测时,MDDMI效果低于单一使用o数据集效果,但与训练效果相当。其余3种疾病中,MDDMI预测效果优于使用单一模态数据。
4 结论
对同一数据集使用不同方法得到的菌群结构以及多样性存在差异,这与OTU、ASV工作原理的不同以及ASV法物种注释覆盖率低有关。数据分析结果表明,虽然ASV法更加精确,但相比OTU法,无法有效识别和分类相似微生物种类,影响低丰度物种分类结果,α多样性较低;疾病预测结果表明,使用OTU数据集训练两种基础模型,在4种疾病数据集中AUC值均高于ASV数据集,表明低丰度属对疾病预测效果的影响较大。通过多模态融合,可得到更全面的视角,提高疾病预测效果。本文提出了一种使用图卷积神经网络融合OTU、ASV以获取物种信息的方法:MDDMI,使用系统发育树构造OTU与ASV异构网络,利用图卷积神经网络获取相关性,考虑显著性分析结果,融合AGP中4种疾病的OTU与ASV数据集进行疾病预测,AUC值普遍高于单模态数据疾病预测结果。
参考文献
[1]TAMBOLI C P, NEUT C, DESREUMAUX P, et al. Dysbiosis ininflammatory bowel disease[J]. Gut, 2004, 53(1): 1-4.
[2]NGUYEN T H, CHEVALEYRE Y, PRIFTI E, et al. Deep learning for metagenomic data: using 2D embeddings and convolutional neural networks[DB/OL]. [2023-08-02]. https://arxiv.org/abs/1712.00244.
[3]NGUYEN T H, PRIFTI E, CHEVALEYRE Y, et al. Disease classification in metagenomics with 2Dembeddings and deep learning[DB/OL]. [2023-08-02]. https://arxiv.org/abs/1806.09046.
[4]ASGARI E, GARAKANI K, MCHARDY A C, et al.MicroPheno: Predicting environments and host phenotypes from 16S rRNA gene sequencing using a k-mer based representation of shallow sub-samples[J]. Bioinformatics, 2018, 34(13): 32-42.
[5]REIMAN D, METWALLY A, SUN J, et al.PopPhy-CNN: A phylogenetic tree embedded architecture for convolutional neural networks to predict host phenotype from metagenomic data[J]. IEEE Journal of Biomedical and Health Informatics, 2020, 24(10): 2993-3001.
[6]NEARING J T, DOUGLAS G M, COMEAU A M, et al. Denoising the denoisers:An independent evaluation of microbiome sequence error-correction approaches[J]. PeerJ, 2018, 6: e5364
[7]CALLAHAN B J, MCMURDIE P J, ROSEN M J, et al. DADA2: High-resolution sample inference from Illumina amplicon data[J]. Nature Methods, 2016, 13(7): 581-583.
[8]BLAXTER M, MANN J, CHAPMAN T, et al. Defining operational taxonomic units using DNA barcode data[J]. Philosophical Transactions of the Royal Society of London, 2005, 360(1462): 1935-1943.
[9]KNIGHT R, VRBANAC A, TAYLOR B C, et al. Best practices foranalysing microbiomes: Nature reviews[J]. Microbiology, 2018, 16(7): 410-422.
[10] PRODAN A, TREMAROLI V, BROLIN H, et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing[J]. PLoS ONE, 2020, 15(1): e0227434.
[11] CARUSO V, SONG X, ASQUITH M, et al. Performance of microbiome sequence inference methods in environments with varying biomass[J]. mSystems, 2019, 4(1): e00163-18.
[12] FORSTER D, LENTENDU G, FILKER S, et al. Improving eDNA-based protist diversity assessments using networks of amplicon sequence variants[J]. Environmental Microbiology, 2019, 21(11): 4109-4124.
[13] MILANESE A, MENDE D R, PAOLI L, et al. Microbial abundance, activity and population genomic profiling with mOTUs2[J]. Nature Communications, 2019, 10(1): 1014.
[14] PRODAN A, TREMAROLI V, BROLIN H, et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing[J]. PLoS ONE, 2020, 15(1): e0227434.
[15] PAUVERT C, BUE M, LAVAL V, et al. Bioinformatics matters: The accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline[J]. Fungal Ecology, 2019, 12(5): 1064.
[16] GRAZIOLI F, SIARHEYEU R, ALQASSEM I, et al. Microbiome-based disease prediction with multimodal variational information bottlenecks[J]. PLoS Computational Biology, 2020, 18(4): e1010050.
[17] MCDONALD D, HYDE E, DEBELIUS J W, et al. Americangut: An open platform for citizen science microbiome research[J]. mSystems, 2018, 3(3): e00031-18.
[18] SHADE A. Diversity is the question, not the answer[J]. The ISME Journal, 2017, 11(1): 1-6.
[19] SU X, XU J, NING K. Parallel-META:Efficient metagenomic data analysis based on high-performance computation[J]. BMC Syst Biol 6 (Suppl 1), 2012, 16(1): 4-8.
[20] ROGNES T, FLOURI T, NICHOLS B, et al. VSEARCH:A versatile open source tool for metagenomics[J]. PeerJ, 2016, 4: e2584.
[21] 钟辉, 刘亚军, 王滨花, 等. 分析方法对细菌群落16S rRNA基因扩增测序分析结果的影响[J]. 生物技术通报, 2022, 38(6): 81-92.
Multimodal Information Fusion of Gut Microbiome for Disease Detection Method
LIU Chang, WU Shun-yao
(College of Computer Science & Technology, Qingdao University, Qingdao 266071, China)
Abstract:
Current methods for analyzing amplicon sequencing data that utilize Operational Taxonomic Units (OTU) or Amplicon Sequence Variants (ASV) can lose multimodal information from various species spectrum construction methods. An analysis was conducted on the differences in community diversity and structure between OTU and ASV datasets across four diseases. An effective approach to integrate OTU and ASV for disease characterization prediction was proposed: MDDMI (Microbiome-based Disease Detection with Multimodal Information). The results indicate that MDDMI is superior to the single-mode data analysis method.
Keywords:
graph convolutional networks; disease prediction; multimodal; gut microbiome
