自噬调控小胶质细胞极化在缺血性脑卒中的研究进展
2024-05-18王方明尚文璇张靖雯吉盈肖李俐涛
王方明 尚文璇 张靖雯 吉盈肖 李俐涛
1河北医科大学研究生院(石家庄 050017);2河北北方学院研究生院(河北张家口 075000);3河北省人民医院神经内科(河北省脑网络与认知障碍疾病重点实验室)(石家庄 050051)
缺血性脑卒中(ischemic stroke,IS)是我国临床最常见的脑血管疾病,约占所有脑血管疾病的75%,致残率极高[1]。目前IS缺乏理想的治疗方法,尽管静脉溶栓治疗及机械取栓手术可改善完全性脑卒中患者的预后,但这些治疗方法存在溶栓时间窗有限、再灌注损伤及再出血并发症等问题,使患者面临继发性脑损伤的高风险。大量研究表明炎症在继发性脑损伤中起到关键的级联损伤作用[1],而且小胶质细胞在参与IS 后炎症反应时,可被激活成两种表型—经典激活型(M1 表型)和替代激活性(M2 表型)[2],其中M1 型和M2 型小胶质细胞分别发挥着促炎和抗炎作用,将小胶质细胞从M1 型转化为M2 型,可以挽救梗死周围的缺血半暗带,促进恢复期神经血管单元重构,所以调节小胶质细胞从M1 到M2 转化是阻止继发性脑损伤的重要途径。研究证实,自噬(autophagy)可调节小胶质细胞的表型转化[3]。本文就自噬对小胶质细胞表型变化的影响在IS 的研究进展进行综述。
1 小胶质细胞极化与自噬概念
1.1 小胶质细胞极化 神经系统炎症反应在IS中发挥重要作用,其中小胶质细胞是中枢神经系统的主要免疫反应细胞。在感染、外伤或缺血后,小胶质细胞可被迅速激活,调节炎症反应。小胶质细胞被激活后可分化为两种表型—M1 型和M2型。其中,M1 型细胞可由细菌脂多糖(LPS)诱导产生,其表面表达CD86、CD68 等,并可以分泌大量炎症因子如肿瘤坏死因子-α(TNF-α)、白细胞介素β(IL-β)、人干扰素-γ(INF-γ)和NO 等;M2 型细胞可由IL-4/IL-10 诱导产生,促使小胶质细胞分泌IL-4、精氨酸酶1(arignase1)、几丁质酶样蛋白质(Ym1)、CD206 及IL-1,前者促进脑缺血后二次损伤,而后者具有脑缺血后的神经保护作用[4]。HU等[5]基于线栓法阻塞C57BL/6J 小鼠左侧大脑中动脉(middle cerebral artery,MCA),通过建立局灶性脑缺血模型,并利用PCR 技术观察到M2 型小胶质细胞标志物(CD206、Arg1、CCL22、Ym1/2、IL-10 和TGF-β)的mRNA 在大脑中动脉栓塞后1 ~ 3 d 开始表达,并于3 ~ 5 d 达到高峰,7 d 开始下降,14 d恢复到损伤前水平。然而,M1 型小胶质细胞相关基因(iNOS、CD11b、CD16、CD32、CD86)的水平从术后第3 天开始逐渐升高,14 d 达到高峰。IS 后小胶质细胞M1/M2 型表达模式的动态变化早期表现出有益的M2 表型,后期转变为有害的M1 表型。调节小胶质细胞从M1 表型向M2 表型转化可能是治疗IS 的关键方法。
1.2 自噬概念 自噬是细胞内分解代谢的一种途径,是受自然调控的过程,可消除细胞内功能失调的成分[6]。其分类方式多样,根据向溶酶体运输货物方式的不同,可分为3 种,巨自噬、微自噬、分子伴侣介导型自噬。巨自噬即由内质网、线粒体、高尔基体或细胞质膜等来源的膜包绕待降解物形成自噬体,然后与溶酶体融合降解相应底物;微自噬指溶酶体膜自身发生凹陷,直接包裹和吞噬细胞内待降解的底物,并在溶酶体内降解;分子伴侣介导的自噬是由分子伴侣蛋白识别并结合带有特定氨基酸序列的可溶性蛋白质,经溶酶体膜上受体转运到溶酶体内,随后被水解酶降解的过程[7]。除此之外,自噬的过程可分为3 个阶段,包括降解的细胞质成分被双层膜吞噬形成自噬小体、自噬小体与溶酶体融合、水解酶催化降解其内容物[8],自噬调节剂可作用于其中某个阶段发挥调节作用。
自噬受多种信号分子调控,这些信号分子包括哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、丝裂原活化蛋白激酶(mitogenactivated protein kinase, MAPK)、5′-AMP 活化蛋白激酶(AMPK)、蛋白激酶B(Akt)、Beclin-1、p53、Rab7 等[9-10]。Beclin1、LC3 和P62 是反映自噬水平的关键指标。Beclin1 与PI3K 结合形成复合物,招募自噬相关蛋白,参与自噬起始过程[11]。LC3 有LC3I 和LC3II 两种形式,由LC3I 向LC3II 转化是自噬体膜形成的关键步骤[12]。P62 蛋白作为自噬降解底物的受体,其累积表示自噬过程的抑制,与自噬水平呈负相关[13]。Beclin1、LC3 水平增多,P62蛋白水平减少代表自噬激活,相反代表自噬抑制[14]。寻找自噬信号靶点,发挥自噬调节作用将是一种潜在治疗方式。
2 自噬调控小胶质细胞极化的研究进展
自噬作为应激条件下的生存机制,在炎症及免疫系统调节方面发挥重要作用[15]。研究发现,在IS 中,自噬抑制剂3-Ma 及mTORC1 抑制剂西罗莫司等干预后,自噬标记物及蛋白发生改变,小胶质细胞极化状态也随之改变[16],提示自噬可介导小胶质细胞的极化,进而影响IS 炎症反应进程,该通路相关基因成为IS 潜在的新治疗靶点,如图1所示。目前研究表示,自噬调节小胶质细胞向两方向极化,探究自噬对小胶质细胞的具体调节靶点,发挥其调节作用,进而促进小胶质细胞向抗炎型转化成为临床获益关键。
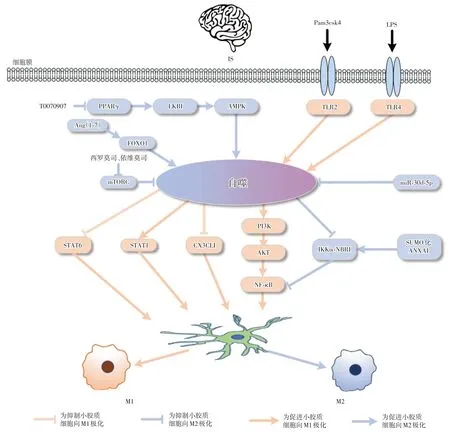
图1 自噬调节小胶质细胞极化过程Fig.1 The process of microglia polarization regulated by autophagy
2.1 自噬促进小胶质细胞向M1 极化 自噬调控小胶质细胞向M1 表型的转化,加剧脑缺血损伤。外泌体具有免疫原性低、稳定性好等特点且易通过血脑屏障,其可调控小胶质细胞自噬作用进而影响极化方向,为目前IS 研究提供新靶点。JIANG 等[16]应用富含MIR-30d-5p 的外泌体处理缺血缺氧的小胶质细胞,检测到自噬相关蛋白Beclin-1 等表达减少,TNF-α 等炎症因子下降,IL-4和IL-10 增加,进一步使用自噬抑制剂3-甲基腺嘌呤(3-Ma)处理小胶质细胞,细胞上清液中促炎因子表达下降,抗炎因子表达增加,证明自噬调控小胶质细胞向M1 极化,且富含MIR-30d-5p 的外泌体可抑制自噬作用。
Toll 样受体(TLR)在中枢神经系统的多种细胞类型中高表达,是机体与环境之间的第一个接触点。因此,特异性调节TLR 的活性或表达可以预防炎症相关疾病的发展。TLR4 依赖性小胶质细胞自噬可通过STAT1/6 通路调控小胶质细胞向M1 极化。QIN 等[17]行双侧颈动脉狭窄术建立慢性脑灌注模型,发现暴露于LPS 的小胶质细胞内自噬体快速积累,转录激活蛋白1(signal transducer and activator of transcription, STAT1)上调及STAT6下调,小胶质细胞M1 型标志物高表达,自噬抑制剂和敲除TLR4 均使小胶质细胞功能从促炎表型转变为抗炎表型,STAT1 的上调和STAT6 的下调被逆转。此外,TLR2 介导的小胶质细胞自噬可以调节其向M1 型转换。MA 等[3]采用肽聚糖(PGN)感染小胶质细胞,联合应用TLR2 拮抗剂CU-CPT22和激动剂Pam3CSK4 干预细胞,发现与PGN 组相比,PGN+Pam3CSK4组自噬标记物LC3II和Beclin-1表达增加,M2 型标志物被抑制,小胶质细胞明显向M1 表型转化,而PGN+CU-CPT22 组观察到相反的结果,自噬增强伴随凋亡增加,凋亡程度与M1表型转换一致。
核因子NF-κB 信号通路与自噬存在密切的相互调控作用,可作为自噬与炎症共同信号分子,在脑组织炎症反应中发挥至关重要的作用。NF-κB介导的小胶质细胞自噬作用,可能成为治疗IS 的重要靶点。XIN 等[18]发现PM2.5 增加M1 型标志物表达,自噬抑制剂3-MA 与PM2.5 共同处理小胶质细胞后,自噬水平降低,M1 型标志物表达减少,M2 型标志物表达增加,表明自噬抑制剂促进小胶质细胞向M2 型极化,进一步实验证实自噬可能通过PI3K/AKT/NF-κB 信号通路调节小胶质细胞向M1 极化。
血清CX3C 趋化因子配体1(CX3CL1)是在神经元上表达的膜结合趋化因子,可以通过与小胶质细胞上的受体CX3CR1 结合来抑制小胶质细胞炎症[19]。神经元自噬通过下调CX3CL1 促进小胶质细胞向M1 极化,HE 等[20]用线栓法建立大脑中动脉阻塞模型,观察到MCAO 干预组血清CX3CL1阳性比例明显降低,Iba-1 和NF-κ B 阳性百分比增加,自噬抑制剂3-MA 处理后,神经元上CX3CL1 表达显著增加,Iba-1 和NF-κ 结果相反。
综上所述,自噬可促进小胶质细胞向促炎型M1 极化,进而加重脑组织损伤程度。外泌体、NFκB 信号分子、Toll 样受体等在未来可能作为调节自噬的靶点,为缺血性脑卒中的治疗提供新思路。
2.2 自噬促进小胶质细胞向M2 极化 自噬在促进小胶质细胞向M2 型转化方面也发挥重要作用。mTOR 可分为两种类型,mTOR 复合物1(mTORC1)和mTOR 复合物2(mTORC2)[21]。mTORC1 信号传导抑制自噬过程,其磷酸化减低可作为自噬激活标志物。LI 等[22]基于线栓法建立MCAO 模型,将mTORC1 抑制剂西罗莫司和依维莫司经口灌入小鼠胃中,小鼠脑内小胶质细胞CD16/32 和Iba-1双染的数量明显减少,通过基因鼠杂交方法阻断mTORC1 相关蛋白,小鼠脑内M1 型小胶质细胞的基因表达下降,M2 型mRNA 的表达显著增加,为说明小胶质细胞自噬调节其向M2 极化提供实验依据。
过氧化物酶体增殖物激活受体γ(PPARγ)是葡萄糖和脂质代谢、细胞器分化及炎症反应的主要调节剂[23],PPARγ 激动剂在应对神经炎症时发挥神经保护作用[24]。JI 等[25]用PPARγ 拮抗剂T0070907 处理LPS 诱导的小胶质细胞,观察到小胶质细胞自噬活动增强,M2 型标志物如CD206 等的表达增加,M1 型标志物如CD86 等的表达成减少趋势。加用LKB1 基因抑制剂根赤壳菌素或对LKB1 基因进行敲除,均可逆转T0070907 的作用进而抑制自噬标志物的表达,促进小胶质细胞向M1极化,说明拮抗PPARγ 可能通过激活LKB1-AMPK信号通路,改善小胶质细胞自噬同时使其向M2 表型极化。
膜联蛋白-A1(ANXA1)是膜联蛋白超家族的37 kDa 成员,参与多种细胞类型中多种细胞功能的调节。中枢神经系统中,ANXA1在小胶质细胞中含量丰富。LI等[26]发现类泛素蛋白修饰分子(small ubiquitin-like modifier, SUMO)修饰的ANXA1 可促进抑制因子激酶IKKα 与自噬受体NBR1 之间的相互作用,增加IKKα 的自噬降解,抑制小胶质细胞中NF-κB 信号通路的激活和促炎介质的产生,导致小胶质细胞向抗炎表型极化。上调小胶质细胞中ANXA1 的SUMO 化可通过小胶质细胞自噬作用调节其向M2 型转化,减少脑梗死体积。
脑肾素-血管紧张素系统(RAS)参与神经炎症,是人体衰老过程的主要特征。大脑ACE2/Ang(1-7)/MasR 轴在自噬与神经炎症之间起关键调节作用。DANG 等[27]发现血管紧张素Ang(1-7)促进了ASC 蛋白的自噬反应,减少ASC 分布,而LC3 分布增加,上述结果可被转入因子FOXO1 的抑制剂AS1842856、氯喹(CQ)完全阻断,提示抑制FOXO1介导的小胶质细胞自噬通路可消除血管紧张素Ang(1-7)的抗炎作用,使其向M1 极化[28],同时自噬促进小胶质细胞向M2 极化。
自噬可促进小胶质细胞向M2 型转化,发挥抗炎作用。其中mTORC1、LKB1-AMPK 信号通路、ANXA1 的SUMO 化及RAS 系统或将成为可干预靶点,为脑卒中的临床治疗提供新方案。
2.3 分析自噬调节小胶质细胞极化 近年来,越来越多研究者发现自噬在小胶质细胞极化中起到双重作用。自噬对小胶质细胞调节的矛盾作用给研究者带来了新的思考。IS 后不同时间点自噬发挥不同作用,早期诱导的适应性自噬有利于神经存活,而长时间或者过度自噬将引起神经损伤。因此,研究者对自噬标志物检测的时间差异可能影响对小胶质细胞极化类型的判断。除此之外,自噬激活剂及抑制剂的选择可能影响实验结果的判断。在实验过程中,3-MA 作为自噬抑制剂被广泛应用,而有研究表明,长时间营养充足的干预环境下,其可发挥自噬促进作用[28],此时选择特异性强的自噬调节剂显得尤为重要。此外实验动物模型类型不同及干预手段差异,甚至实验动物种类及性别不同可能影响实验结果,造成自噬对小胶质细胞极化表型影响的矛盾关系。自噬对小胶质细胞极化的调节机制尚未被完全阐明,需要进一步深入研究。
3 自噬调节剂的应用
目前认为,自噬在脑卒中中扮演着“双刃剑”的作用,其对小胶质细胞极化的调节成双向性,因此,通过自噬调节剂调节自噬平衡可成为IS 防治的关键。自噬受复杂的信号网络调控,其上游调节因子主要为TOR 复合物和AMPK 共同调节。自噬调节剂种类多样,其可经PI3K、AMPK-mTOR 及MAPK 等信号介导发挥调节作用,按照其功能可分为自噬激活剂及自噬抑制剂两种,如图2 所示。自噬调节剂的应用可成为治疗IS 的一个方向,对其选用以及研究开发显得尤为重要。

图2 自噬调节剂的应用Fig.2 Applications of autophagy regulators
雷帕霉素主要通过抑制mTORC1,激活ULK1复合物,促进自噬起始[29],进而对缺血后神经血管单元进行调控,促进神经保护作用,目前临床应用有限[30]。异氟醚是一种常见的气态麻醉剂,常用于神经外科的镇静和麻醉,ZHAI 等[31]发现异氟醚通过激活AMPK/ULK1 信号通路增强自噬,进一步抑制NLRP3 炎症小体炎症因子的释放,从而改善CIRI 大鼠的神经功能和认知障碍,对大脑发挥保护作用。然而,对于新生儿和老年患者,异氟醚有神经毒性作用[32],其在IS 方向的临床应用受限,具体机制仍需要摸索。HECKMANN 等[33]将Ⅲ类PI3K 抑制剂3-甲基腺嘌呤(3-MA)加入3T3-L1 脂肪细胞细胞培养,观察到其显著降低细胞内LC3-II 的水平,抑制自噬作用。3-MA 抑制缺血诱导的LC3-II 表达的同时,也可通过抑制自噬泡延伸进而抑制小胶质细胞自噬[34],从而抑制炎症反应,3-MA 在脑缺血后作用机制及效果尚不明确,目前无相关临床应用研究[35]。黄芩素是一种从黄芩根部提取的生物活性成分,用于抑制癌细胞增值,减少糖尿病及糖尿病并发症等[36],研究表明,其可通过PI3K/Akt/mTOR 信号通路抑制自噬过程,并通过NF-kB 和MAPK 信号通路抑制小胶质细胞的M1转化和神经炎症[37]。另外,芬戈莫德主要用于多发性硬化治疗[38],可作用在自噬起始阶段,通过激活mTOR/p70S6K 通路,抑制自噬[39];长春花碱抑制自噬体与溶酶体的结合,抑制自噬,目前无IS 相关临床应用;氯喹用于神经胶质瘤的治疗[40],通过抑制溶酶体酸化进而抑制自噬过程[41]。自噬调节剂为IS 治疗提供希望,但目前临床应用有限,缺乏研究数据,寻找自噬调节小胶质细胞极化靶点,应用自噬调节剂调节小胶质细胞向M2 型转化将是下一步亟待解决的问题。
4 IS 后自噬与炎症相互作用
自噬通过调控小胶质细胞极化发挥促炎或抗炎作用,同时炎症也影响自噬过程,如图3 所示。细胞因子和活性氧(ROS)可通过改变细胞内环境调节自噬,其中Th1 家族细胞因子(IL-2、TNF-α等)诱导自噬产生,Th2 家族细胞因子(IL-4、IL-5、IL-6、IL-10 等)和抗炎细胞因子抑制自噬作用[42]。病原相关分子模式(PAMP)或损伤相关分子模式(DAMP)可通过识别Toll 样受体(TLR)、NOD 样受体(NLR)激发炎症信号并促进自噬激活[43]。适度自噬通过消除炎症蛋白和促炎细胞因子来有效抗炎,过度自噬会促进炎症反应[44]。自噬与炎症相互联系,互相制衡,与此同时NF-κB、PPARγ、STAT 家族及TLR、NLR 等发生动态变化,影响小胶质细胞表型转化方向[45]。脑缺血后自噬与炎症动态平衡被破坏,小胶质细胞作为其中主要参与细胞发挥着重要调节作用,探索IS 后自噬对小胶质细胞极化调节作用,对减轻缺血后脑损伤有重要价值。

图3 炎症、自噬与小胶质细胞极化相互作用关系Fig.3 The interaction between inflammation, autophagy and microglia polarization
5 小结
小胶质细胞表型改变是IS 的潜在致病机制,且研究表明自噬可通过调控小胶质细胞极化,参与IS 诱导的炎症反应过程。本文概述了自噬对小胶质细胞表型转化的调控作用,对自噬调节剂进行了总结,并讨论了自噬与炎症相互作用关系。促进小胶质细胞由M1 型向M2 型转化,对发挥小胶质细胞的神经保护作用,从而治疗IS 有重要意义。然而,截止目前自噬在小胶质细胞极化的分子调节机制尚不明确,缺乏对IS 病理学的相关研究,而且现有研究存在不同结论。自噬对IS 过程中小胶质细胞表型转化机制有待进一步研究。
【Author contributions】WANG Fangming designed and wrote the article; SHANG Wenxuan and ZHANG Jingwen collected the literatures; JI Yingxiao revised the article; LI Litao was responsible for the quality control and revision of the article and was in charge of the overall supervision and management of the article. All authors read and approved the final manuscript as submitted.
【Conflict of interest】The authors declare no conflict of interest.
