糖尿病致栓性因素的研究进展
2024-05-06王宗奎李长清
龙 茜 王宗奎 李长清 张 容
糖尿病是一种以血糖升高为特征的慢性代谢性疾病,根据有无胰岛素绝对缺乏可以分为1型糖尿病(type 1 diabetes mellitus,T1DM)和2型糖尿病(type 2 diabetes mellitus,T2DM)[1]。据估计,2021年全球20~79岁糖尿病患病率估计为10.5%(5.366亿人),2045年上升至12.2%(7.832亿人)[2]。糖尿病(diabetes mellitus,DM)患者往往伴随着多种并发症,其中心血管疾病是DM患者发病和死亡的主要原因,而血液高凝和血栓前状态是DM心血管并发症首要病理生理过程[3]。
血栓前状态是动脉粥样硬化血栓形成过程中的关键环节。与健康人群比较,DM患者具有更高的凝血功能异常和血栓前状态风险[4]。T1DM和T2DM大都存在凝血功能异常和血栓前状态,主要表现为凝血、抗凝和纤溶相关蛋白浓度和活性的变化(表1)[5]。凝血、抗凝和纤溶相关因子的改变在DM血管并发症的病理生理过程中发挥重要作用。但到目前为止,代谢紊乱导致DM止凝血功能异常的潜在机制尚未完全明确[6]。本文就导致DM凝血异常和血栓前状态的因素进行综述,以期为DM血栓和心血管并发症的防治提供参考。
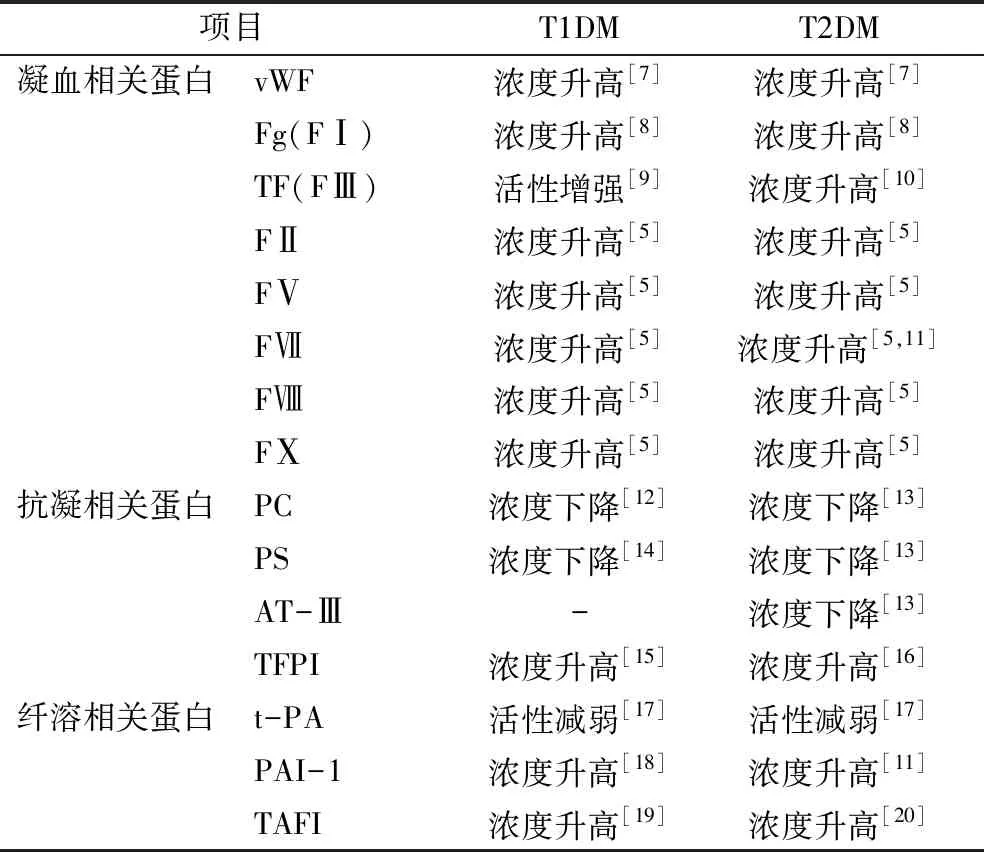
表1 凝血抗凝纤溶相关蛋白的变化
一、促凝因子变化
目前关于DM凝血功能异常已有许多研究,结果显示,高血糖、氧化应激和遗传因素共同作用引起凝血相关蛋白如凝血因子Ⅰ(factor Ⅰ, FⅠ,又称纤维蛋白原,Fg)、凝血因子Ⅲ(factorⅢ, FⅢ,又称组织因子,TF)和凝血因子Ⅶ(factor Ⅶ,FⅦ)等数量和结构的改变,最终导致凝血异常。如图1所示,高血糖引起Fg、TF、FⅦ和vWF血浆水平升高的具体机制已有报道,但关于FⅧ升高机制尚存在不同研究结果,具体机制还有待于进一步探讨。
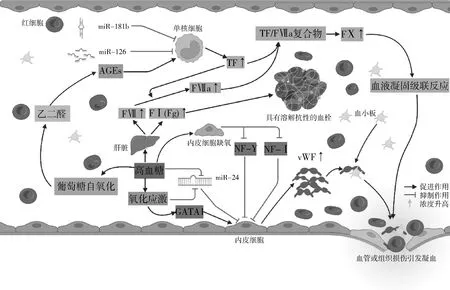
图1 DM凝血相关蛋白变化机制
1.凝血因子变化:凝血因子是控制可溶性血浆Fg向纤维蛋白转化的蛋白质系统,当体内凝血因子水平升高,血液呈现血栓前状态和凝血增强。如图1所示,在T1DM和T2DM患者中Fg、FⅦ等凝血因子血浆浓度都有不同程度升高从而引发凝血功能异常。
FⅦ是外源性凝血途径的始动因子之一,当血管壁受损时,病变处TF激活FⅦ,形成FⅦa/TF复合物,随着FⅦ消耗,DM患者FⅦ反应性增加[21]。Ceriello等[21]研究发现,诱导高血糖能够增加DM患者和正常受试者FⅦ水平,并且当DM患者恢复正常血糖时,FⅦ水平也恢复到正常范围。说明在DM患者中,循环血糖很可能是调控FⅦ的水平的原因之一。Edén等[22]研究发现,人原代脂肪细胞可以表达具有活性的TF和FⅦ,脂肪细胞表面形成的TF/FⅦa复合物可以激活FⅩ,启动凝血。也有研究表明,在DM大血管病变或肥胖的DM患者中FⅦ水平升高尤为明显, FⅦ可能参与了糖尿病血管病变的发生、发展并与DM及其相关并发症密切相关[11]。提示肥胖DM患者血浆FⅦ水平升高可能是脂肪细胞表达具有活性的TF和FⅦ所导致。以上研究提示DM患者FⅦ水平升高可能是多因素共同作用的结果,至于各因素有无相互作用尚有待于进一步探讨。
2. vWF水平升高:血管性血友病因子(Von Willebrand factor,vWF)是一种介导血小板沿内皮黏附的糖蛋白,血管内皮受损时,它通过将血小板锚定到受损血管的内皮下基质来促进血小板黏附,并保护FⅧ免受蛋白水解酶降解。Mattia等[7]研究发现,DM患者的血浆vWF水平显著高于健康受试者。T2DM合并心血管疾病患者血浆vWF和凝血酶的浓度也显著高于不合并心血管疾病的患者[23]。在有血糖异常和其他心血管危险因素的人群中,与标准降糖治疗比较,基础胰岛素治疗不能改善纤溶标志物或vWF水平[24]。
近年来,有研究者就DM中vWF水平升高的作用机制进行了研究。研究发现,microRNA-24 (miR-24)可以通过与vWF分泌调节因子(FURIN和H1组胺受体)结合而抑制内皮细胞分泌vWF,而高血糖和氧化应激却能通过下调miR-24表达促进vWF的分泌(图1)[25]。此外,高血糖诱导的内皮细胞缺氧会下调vWF启动子的两个抑制分子NF-I和NF-Y的水平,从而增加vWF的分泌(图1)[25]。GATA家族是内皮细胞中vWF表达的转录因子,高血糖诱导的氧化应激通过GATA1转录因子上调内皮细胞中vWF的表达(图1)[26]。这为调节内皮细胞中vWF的表达和改善DM高凝状态提供了潜在治疗靶点。高血糖和氧化应激可以通过不同通路影响vWF的分泌,对DM患者加强血糖管理是降低vWF水平和血栓形成最基础的方法。此外,研究还发现,内皮细胞Sirt1/FoxO1通路被激活会抑制vWF的分泌,但该作用可以被自噬抑制剂消除[27]。说明内皮细胞可能通过Sirt1/FoxO1通路增强自噬而减少vWF的释放,从而减少血栓形成风险。但DM患者是否存在自噬抑制而导致vWF分泌增多值得进一步探究。
3. TF表达增强:TF是FⅦ和活化的FⅦ(activated FⅦ,FⅦa)的受体和辅因子,催化FⅨ和FⅩ的活化。TF-FⅦa复合物是外源性凝血途径的主要激活剂,如图2,在血管损伤或组织创伤后,TF-FⅦa复合体通过激活FⅩ触发血液凝固的级联反应[28]。Soma等[10]通过临床研究发现,无论是否伴有心血管疾病,T2DM患者血浆TF水平大约是健康对照组的2.5倍。在T1DM患者中,循环组织因子促凝活性(TF-PCA)增强, FⅦa水平升高[9]。T2DM患者普遍存在高血糖和高胰岛素血症,而高血糖和高胰岛素血症可以单独或联合增加T2DM患者TF浓度和TF-PCA,说明T1DM和T2DM患者发生心血管事件风险都较高[29]。
晚期糖基化终末产物(advanced glycation end-products, AGEs)是由还原糖与核酸、蛋白质或脂类通过非酶促反应后进一步重排,产生的一系列稳定、不可逆的化合物[30]。在过去的几十年里,越来越多的研究表明,AGEs参与了DM的病理生理过程。在DM患者中,高血糖引发的葡萄糖蓄积导致葡萄糖自氧化,从而形成AGEs的前体——乙二醛[31]。AGEs可以在mRNA水平上诱导单核细胞表达TF,但添加抗氧化剂可以降低该途径下TF的表达,说明AGEs诱导的TF表达很可能是通过氧化应激途径实现的[32]。在T2DM中,microRNA-181b(miR-181b)会减少单核细TF的产生并且血浆miR-181b水平与TF活性、D-二聚体水平以及血管炎症标志物等致栓性因素负相关[33]。循环microRNA-126(miR-126)通过调节转录后TF的表达,影响DM的凝血纤溶平衡,从而发挥抗血栓作用[34]。这些研究结果都在一定程度上解释了DM血浆TF升高的原因,但其中是否涉及其他机制还需进一步研究。
4.Fg水平升高:Fg是体内最丰富的凝血因子,在凝血过程中,Fg通过凝血酶介导的蛋白水解作用分解为纤维蛋白,产生中间原纤维,形成具有生化和机械稳定性的纤维蛋白网。如表1,T1DM和T2DM患者Fg水平显著升高[8]。
血糖控制不佳的DM患者体内的高血糖可引起Fg糖基化,导致Fg分子结构和功能的改变,并有助于形成稳定致密的血栓,且与非糖尿病血栓比较,糖尿病血栓对纤维蛋白溶解抗性更强[35]。高Fg血症可能是T2DM微血管和大血管血栓风险增加的机制之一[36]。BβArg448Lys是Fg常见的遗传多态性之一,Greenhalgh等[37]研究发现,相较于其他基因型血栓,Bβ448Lys基因型血栓溶解时间更长,血栓结构也更加紧密,这些结果均提示除高血糖外,遗传因素也是增加DM血栓形成和溶解抗性风险的原因之一。
二、抗凝因子变化
血液中的抗凝因子主要分为3大类:抗凝血酶-Ⅲ (antithrombin-Ⅲ, AT-Ⅲ)、蛋白C系统和和组织因子途径抑制物(tissue factor pathway inhibitor,TFPI)。在T1DM和T2DM中,三大抗凝因子均发生了一定变化。
1.AT-Ⅲ减少: AT-Ⅲ是一种主要的内源性抗凝剂,其缺乏会导致严重的血栓形成倾向。研究发现,T2DM患者AT-Ⅲ血浆浓度降低并且与血糖水平相关,高血糖副产物乙二醛通过与AT-Ⅲ活性位点(精氨酸-393残基)共价结合导致AT-Ⅲ失活,从而降低AT-Ⅲ对FⅡa和FⅩa的抑制[13, 25]。也有研究表明,高血糖通过引起内质网氧化应激使AT-Ⅲ和α1-抗胰蛋白酶滞留和聚集在细胞内,导致DM小鼠循环AT-Ⅲ缺乏[38]。并且DM患者在接受胰岛素治疗改善血糖水平之后,AT-Ⅲ的功能也有显著改善[39]。Caseiro等通过对T1DM患者尿液进行蛋白质组学分析,发现其中含有AT-Ⅲ[40]。这也从侧面说明了T1DM血浆中AT-Ⅲ丢失过多,但关于AT-Ⅲ在T1DM血浆的变化和机制还未见报道。
2.蛋白C系统紊乱:蛋白C系统是在凝血过程中具有抗凝作用的血浆蛋白系统,包括PC、PS、血栓调节蛋白及活化的蛋白C抑制物。在T1DM和T2DM中,PC和PS的血浆浓度均低于非糖尿病患者,且PC水平与血糖水平呈负相关[5,14]。Vukovich等[12]研究发现,T1DM患者循环凝血酶对PC的激活增强,导致血浆中PC清除增加。同样,在血糖控制不佳的T2DM患者中,PC、PS和AT-Ⅲ的活性和浓度都降低[13]。总之,血糖水平是影响PC和PS等抗凝相关蛋白的最主要因素。但至今为止还没有研究阐明血糖水平对PC和PS血浆水平和活性的影响机制,相关研究有待于进一步开展。
3.TFPI增多:TFPI是一种凝血抑制因子,会抑制凝血酶的生成,尤其是在凝血酶生成的早期阶段。儿童和青少年T1DM患者的TFPI水平显著较高且与血糖控制相关,高血糖可通过激活内皮细胞增加TFPI水平[15]。T2DM患者TFPI水平显著升高,并且同样与血糖控制相关[16]。即使T1DM和T2DM都存在TFPI水平升高,但其血糖升高同时也伴随着TF的活性增加,TFPI水平的升高并不能完全代偿TF活性增加所带来的促凝血状态,患者仍有较高的血栓形成风险[41]。聚腺苷二磷酸核糖聚合酶-1[poly(ADP-ribose)polymerase-1,PARP-1]是DNA损伤的传感器,具有识别DNA损伤的功能,并通过招募DNA修复机制到损伤部位来促进DNA修复[42]。动脉粥样硬化和动脉粥样硬化性钙化与血栓形成密切相关。Stat1作为正性转录因子可以直接结合启动子Runx2导致DM动脉粥样硬化性钙化,而PARP-1能激活Stat1转录介导的Runx2表达,从而促进DM动脉粥样硬化性钙化的形成[43]。Wang等[44]研究发现,PARP-1缺陷小鼠TFPI2活性明显增强,最后抑制高血糖诱导的内膜增生,说明血管平滑肌增殖和迁移可以被TFPI2抑制。而血管平滑肌增殖和迁移导致的血管重塑是动脉粥样硬化血栓形成的重要原因。因此,PARP-1有可能成为预防DM患者血栓形成的有效靶点。
三、纤溶系统紊乱
纤维蛋白溶解系统也称为纤溶酶原-纤溶酶系统,由纤溶酶及其前体纤溶酶原组成。纤溶酶原激活剂有两种,组织型纤溶酶原激活剂(t-PA)和尿激酶型纤溶酶原激活剂(u-PA)。纤溶酶抑制剂由α2-纤溶酶抑制剂和α2抗纤溶酶组成。高血糖和胰岛素抵抗等通过多种途径影响DM患者的纤溶系统。
1. 纤溶酶和纤溶酶抑制剂紊乱:DM高血糖会导致纤溶酶原糖基化,从而损害蛋白质功能[17]。T1DM患者纤溶酶原转化为纤溶酶的速率降低,导致纤溶功能受损,但改善血糖控制可在一定程度上恢复纤溶酶原向纤溶酶的转化及相关酶的活性[17]。并且适度降低血糖就足以显著改善纤溶酶的活性[45]。表明血糖水平改善在降低糖尿病血栓形成风险中至关重要。研究表明,DM患者的血浆纤溶酶抑制剂水平升高,纤溶酶抑制剂水平与HbA1c水平呈正相关[46]。在T1DM和T2DM患者中,FⅫ催化的纤溶酶抑制剂交联到纤维蛋白网络中增多,增加纤维蛋白网络的裂解抗性,导致低纤溶状态的形成[47]。近年来一项研究表明,与男性T2DM患者比较,纤维蛋白中α2-抗纤溶酶掺入增加可能是女性T2DM患者的纤溶功能受损的部分原因[48]。说明纤溶酶抑制剂渗入纤维蛋白网络可能存在性别差异,但目前仅进行了体外研究,尚不清楚造成此差异的具体机制。
2.t-PA和PAI-1紊乱:t-PA是一种丝氨酸蛋白酶,通过将纤溶酶原转化为纤溶酶,从而启动纤溶过程。在DM患者中,t-PA与HbA1c呈负相关,血糖水平升高会抑制t-PA的活性,引起血浆纤溶酶水平降低[17]。组织型纤溶酶原激活物抑制剂-1 (PAI-1)是最强大的抗纤溶蛋白之一,可与t-PA或u-PA结合,减少纤溶酶的生成,使血栓溶解时间延长,在糖尿病和胰岛素抵抗状态下PAI-1水平升高[49]。PAI-1也可以通过抑制血管壁释放t-PA,降低游离t-PA水平和活性,导致纤维蛋白溶解过程受损[50]。高血糖通过影响两个相邻的Sp1位点增加PAI-1在体外血管平滑肌细胞上的表达,从而提高PAI-1的浓度和活性。在胰岛素存在的情况下高血糖通过增强氧化应激刺激人肝细胞中转录因子核因子κB活性升高,最终增强Fg和PAI-1等凝血主要调节蛋白的基因转录。
3. TAFI紊乱:TAFI在凝血平衡和纤溶系统中具有重要的调节作用,活化的TAFI可通过从部分降解的纤维蛋白羧基末端移除赖氨酸残基从而可有效地抑制纤维蛋白溶解。T1DM患者血浆TAFI水平显著高于健康人群可能是T1DM患者血管内皮损伤的机制之一[19]。T2DM患者血浆TAFI水平也同样升高且与发病风险相关,因此TAFI也可以作为T2DM诊断的潜在标志物[20]。高血糖症和胰岛素抵抗导致TAFI水平升高,从而导致低纤溶状态,而血糖正常时可逆转TAFI的变化[17]。表明了血糖控制在缓解DM纤溶功能障碍中的重要性。
综上所述,DM血栓前状态是多系统多因素共同作用的结果,凝血抗凝和纤溶系统失衡都在其发生、发展过程中起重要作用。在凝血、抗凝和纤溶系统中,高血糖和氧化应激是导致致栓性因素出现或增多的核心原因。虽然已发现众多导致DM血栓前状态的因素,但是其进一步机制尚未明确。因此,还需进一步借助体内体外实验去探究高血糖和氧化应激等代谢因素对DM凝血的影响,以便为DM抗血栓治疗提供准确的方向和治疗靶点。
利益冲突声明:所有作者均声明不存在利益冲突。
