3d过渡金属软X射线吸收谱学的理论计算进展
2024-04-29樊亚磊周靖胡志伟王建强张林娟
樊亚磊 周靖 胡志伟 王建强 张林娟
1(中国科学院上海应用物理研究所 上海 201800)
2(马克斯-普朗克固体化学物理研究所 德国 德累斯顿 01187)
3(中国科学院大学 北京 100049)
过渡金属(Transition Metal,TM)化合物具有丰富的物理化学性质,如金属-绝缘体转变、庞磁电阻、高温超导、电催化等[1],其大多源于TM离子的电荷、轨道和自旋自由度之间的强相互作用[2-4]。由于3d TM中不仅存在3d电子间的相互作用,而且3d轨道与配体轨道也存在杂化作用,所以其电子结构非常复杂。以Co离子为例,不仅具有+2、+3、+4等多种氧化态,还存在着多种自旋态:Co3+和Co4+离子可处于高自旋(High-Spin,HS)(S=2;S=5/2)、低自旋(Low-Spin,LS)(S=0;S=1/2)、中间自旋(Intermediate Spin,IS)(S=1;S=3/2)状态等。精确确定TM离子的价态、轨道占据和自旋状态等电子结构信息有助于从微观层面上理解材料所表现出来的复杂物理化学性质,对于针对性地设计和开发新材料有着重要的推动作用,在材料、能源、化学等领域日益受到重视。但遗憾的是,这些复杂电子结构的探测与解析目前仍存在很高的难度。
基于同步辐射的软X射线吸收谱(soft X-ray Absorption Spectroscopy,sXAS)具有元素选择性,对TM的价态、自旋态、轨道排列等电子结构极其敏感,因此是研究TM基化合物电子结构的重要手段之一[5]。以TM-L2,3边为例,sXAS可以直接获取3d电子结构信息,光谱的多重结构特征与TM 3d-3d、2p-3d库仑和交换相互作用、局域晶体场以及与配体O-2p杂化等信息直接相关,因此被广泛应用于3d电子结构研究中。
对于3d TM的sXAS光谱,由于芯态空穴和价电子波函数之间的重叠以及3d电子之间存在着较强的电子相互作用,使得3d电子轨道劈裂复杂,无法使用单电子近似方法对其进行解析。基于配体场理论的多重态计算方法包含完整的原子多重态理论,如原子内库仑相互作用和自旋轨道耦合作用,以及固体的局域效应,包括晶体场和TM离子与配体之间的杂化等[6--9],因此可以很好地解析sXAS光谱。此外,虽然C、N、O等配体的K边也在sXAS的波段范围,但他们的电子结构可以通过基于密度泛函理论(Density Functional Theory,DFT)的能带计算很好解释。
虽然sXAS已经在物理、化学等领域有着广泛应用,但与材料性质密切相关的电子结构信息由于多电子相互作用以及晶体场相互作用的复杂,使得sXAS的理论计算发展滞后于其应用。本文详细梳理了sXAS谱学理论解析方法的基本原理,特别阐明了微观电子结构与TM基化合物宏观性能之间的关系。
1 XAS计算的基本原理
对3d TM而言,K边的跃迁(1s→np)可以通过基于单电子近似的密度泛函理论等计算方法很好地解释。然而,对于3d TM的L2,3边XAS(2p→nd)来说,由于TM芯空穴和3d价电子波函数之间以及3d电子之间的强相互作用,多重态效应对除了3d0和3d9外的所有电子构型都及其重要,即使是3d1构型也存在显著的多重态效应,对sXAS的谱形有着不可忽视的影响[10],因此无法用单电子模型理论很好地描述。在过去的几十年中,基于多重态理论发展的计算方法已经能够成功对L2,3边进行定量描述,能够解释其中3d电子的多重态效应。基于配体场理论的组态相互作用模型(Configuration-Interaction Cluster Model,CICM)计算与实验存在很好的一致性,被广泛用于研究TM的各种物理性质。
1.1 L2,3边的原子多重态理论
在X射线吸收过程中,具有足够能量的光子被原子吸收,导致芯电子跃迁到费米能级以上的空态,对于具有2p63dn初态构型的TM离子,其终态构型应为2p53dn+1。由于2p自旋轨道的耦合作用,芯空穴可以位于6个不同的轨道,这些轨道会劈裂为4个j =3/2的轨道和2个j = 1/2的轨道。例如,对于2p63d9的初态构型,其终态2p53d10构型可以分为两组6个终态。但是对于n < 9的初态构型,还必须考虑3d轨道上的空穴,该空穴可以位于10个不同的轨道上,导致存在更多的终态构型,这种效应称为多重态效应。
CICM理论考虑的自由原子的总哈密顿量可以由下式表示:
式中包含了原子中N个电子的动能(第一项),电子与原子核的库仑相互作用(第二项),电子与电子之间的排斥(第三项)和每个电子的自旋-轨道耦合(第四项)。
在进行计算时,可以根据不同组态的轨道矩L、自旋矩S和总角动量J来分类不同的项。不同项的相对能量可以由有效的电子-电子相互作用和自旋-轨道耦合决定。其中,原子内的有效库仑相互作用的矩阵元素可以表示为:
其中:Fk(fk)和Gk(gk)分别是2p-3d的库仑项和交换项的多极相互作用的径向(角度)部分的Slater-Condon参数[11]。对于3d TM来说,原子内的电子-电子相互作用与分开L2边与L3边的2p自旋-轨道耦合具有相同的数量级。
在2p X射线吸收过程的末态,主要的相互作用是芯空穴的自旋轨道耦合。3d TM的2p轨道自旋轨道耦合作用使得L边大体上被分成L3和L2两部分。以具有Oh对称性结构的Ti4+为例,在不考虑任何相互作用的情况下,sXAS光谱仅包含一个单峰,如图1(a-1)所示。如果计算时考虑2p轨道的自旋轨道耦合作用,L边将被分成L3和L2两部分,如图1(a-2)所示[12-13]。进一步考虑多重态效应时,会导致卫星峰的出现,如图1(a-4)所示。
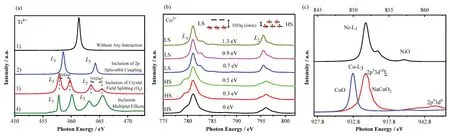
图1 (a) 考虑不同电子相互作用情况下的Ti4+理论光谱:1) 无相互作用,2) 存在2p自旋-轨道耦合,3) 存在晶体场相互作用和4) 包含多重态效应;(b) 具有Oh对称性的Co3+取不同10 Dq时的理论光谱;(c) NaCuO2(Cu3+)、CuO(Cu2+)和NiO(Ni2+)的L3边实验光谱[19]Fig.1 (a) Theoretical spectrum of Ti4+ under different electron interactions: 1) no interaction, 2) including 2p spin-orbit coupling, 3)including crystal field interaction, and 4) including the multiplet effect; (b) The theoretical spectrum of Co3+ with Oh symmetry at different 10 Dq; (c) Cu L3 edge experimental spectra of NaCuO2(Cu3+) and CuO(Cu2+), and Ni L3 edge experimental spectra of NiO(Ni2+)[19]
1.2 晶体场作用
原子多重态理论能够精确地描述稀土元素的3d和4d吸收谱[14],但是由于3d TM离子配体的影响较强,为了考虑TM离子周围环境的影响,Bethe[15]和van Vleck[16-17]自20世纪30年代以来发展了晶体场理论,在晶体场理论中,离子势能项来源于TM离子周围的原子。晶体场理论模型的出发点是将TM离子近似为一个被周围电荷分布所包围的孤立原子。新的哈密顿量由原子哈密顿量和晶体场项组成,其中晶体场项可以视为原子哈密顿量的微扰。晶体场势可以写为:
式中:ρ为周围离子的电荷密度;R表示相对于中心原子的坐标。对于几种常见晶体结构,电子的轨道由局域对称性决定。例如,在八面体(Oh)对称性结构中,d轨道劈裂为双重简并的eg轨道和三重简并的t2g轨道。eg和t2g轨道之间的能量劈裂通常称为晶体场劈裂能10 Dq。双重简并的eg轨道由TM离子指向配体方向的轨道组成,它们与配体之间存在强相互作用。在Ti4+结构中,晶体场的作用会使L2,3边劈裂为eg峰和t2g峰,eg峰和t2g峰之间的能量差为10 Dq,如图1(a-3)所示。在四方(D4h)和三角(D3d)对称性结构中,晶体场劈裂方式与Oh相似,D4h的eg轨道会进一步劈裂为两个非简并的a1g轨道和b1g轨道,t2g轨道也会进一步劈裂为一个b2g轨道和双重简并的eg轨道;而在D3d下,t2g轨道则会进一步劈裂为一个a1g轨道和双重简并的eπg轨道,如图2所示。
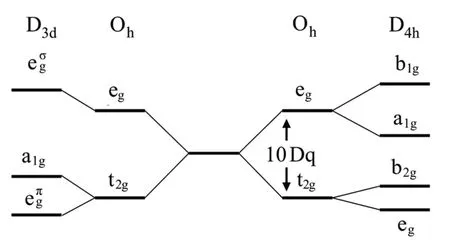
图2 三角对称(D3d)、八面体对称(Oh)、四方对称(D4h)性下的d能级劈裂Fig.2 Energy-level diagram for the d shell in trigonal (D3d),octahedral (Oh), and tetragonal symmetry (D4h)
晶体场劈裂能的大小也会影响体系的自旋态。例如,对于具有3d6构型的Co3+,不同自旋态的总能量应为:EHS= -10Jex- 24 Dq、ELS= -6Jex-4 Dq,其中Jex为交换项。因此,当晶体场劈裂能变小时,Co3+的HS态相比LS态更有利,理论光谱如图1(b)所示。计算时采用的其他参数分别为:Udd= 5.5 eV、Udp=7.0 eV、Δ= 2.0 eV、Tpp= 0.5 eV、 Salter Integrals =80%。当从高自旋态转变为低自旋态时,10 Dq值由0.5 eV增加到0.7 eV。
后来发展起来的配体场理论进一步考虑了TM离子和周围配体之间的共价杂化作用[18]。对于3d TM,TM离子和配体之间的杂化使得实际的波函数难以用单一的组态进行描述。因此,配体场理论进一步考虑了中心离子和周围配体之间的成键作用,成键作用可以看作是一种额外的原子组态间的相互作用。特别是对于高价态的后3d TM离子,这种中心离子和配体之间的电荷转移可以被描述为组态相互作用,并用一系列组态的线性组合来描述波函数。
1.3 组态相互作用模型
组态相互作用模型起源于TM-3d和O-2p电子间的强共价相互作用,并导致O-2p到TM-3d的电荷转移。例如,当考虑TM-3d和O-2p之间的共价相互作用时,Mn3+、Fe3+、Co3+、Ni3+、Cu3+的基态构型不再为3dn(n分别为4、5、6、7、8、9)。电子在组态相互作用的计算中,初态和末态的波函数可以分别用一系列组态的线性叠加来表示。初态可以表示为2p63dn,。终态则可以表示为,其中表示O-2p上的空穴。在目前的计算中,一般只考虑能量最低的两到三种组态。因此,初态的波函数可以表示为:
终态的波函数可以表示为:
式中:i表示初态;f表示末态。
例如,NaCuO2(Cu3+)和NiO(Ni2+)具有同样的电子构型(3d8)。但对于高价态的Cu3+,强共价相互作用导致存在多种末态构型,例如,其主峰和卫星峰处的末态构型分别为和2p53d9,造成了NaCuO2和NiO谱形的差异,如图1(c)所示[19]。
2 sXAS谱学计算软件发展现状
对于稀土元素,由于其轨道局域性较强,晶体场的强度较弱,仅考虑原子多重谱线就能够准确描述,所以稀土元素sXAS光谱的计算是首先发展起来的。相对来说,由于3d过渡金属d轨道与周围配体存在强的相互作用,在计算时必须要考虑配体场的影响。经过多年发展,目前对3d过渡金属sXAS光谱的计算软件也有多种,其中基于组态相互作用模型开发的代码有CTM4XAS[20]、XTLS 9.0[21],以及最近发展起来的Quanty[22]等。
CTM4XAS程序是在Theo Thole开发的电荷转移多重模型基础上,经de Groot和Ogasawara等[23]完善发展而来。该计算方法主要考虑的有:1)芯能级与价电子之间的自旋-轨道耦合;2)原子的多重态效应;3)强关联体系中电荷转移作用的影响。CTM4XAS是目前应用最为广泛的计算软件之一,拥有强大的功能,可以计算TM体系的XAS、XPS和XES等光谱。CTM程序本身也可以用于计算俄歇、共振光电发射等光谱。然而,目前开放版本中考虑的组态较少,对于高价3d过渡金属体系可能会存在一定误差,而且程序所能考虑的对称性有限(目前仅包括Oh、Td等),对于低对称性结构体系难以应对。CTM4XAS程序获取网址为:https://anorg.chem.uu.nl/CTM4XAS/software.html。
XTLS 9.0代码由Tanaka等[21]开发,该计算软件具有计算自由度大的特点。在计算时可以自主定义轨道、组态、对称性等,适用于物理、化学、材料等多种领域,可以实现XAS、XPS等多种光谱的拟合。但是目前还没有可视化的界面,且需要给定的计算参数较多,使得该计算软件的使用较为复杂。XTLS 9.0代码获取网址为:https://github.com/XTLS/。
Quanty程序主要由Haverkort[22]开发,目前仍处于起步阶段,但发展迅速,该代码的理论基础源于计算TM和稀土化合物的芯能级光谱。Quanty融合了密度重整化理论和量子化学思想,可以用来计算各种不同的芯能级光谱,Quanty的灵活性允许人们为不同的材料选择不同的近似。Quanty可以计算X射线吸收、光电发射、X射线发射、共振非弹性X射线散射光谱等[24-26]。Quanty代码获取网址为:https://quanty.org/。
因为基于组态相互作用模型的程序在计算时都需要引入较多的非结构参数,计算过程也较为复杂,目前有部分科研人员尝试发展一种无参数的计算方法。例如,Ikeno等[27]发展了一种基于相对论组态相互作用理论的计算,它是一种多重态结构的无参数从头计算方法。这种方法是在单电子模型的基础上进行修正的,将单电子狄拉克方程的本征函数作为CI计算的基态,其中包括了相对论效应和配体场效应的影响。对于许多TM化合物,sXAS光谱的形状、化学位移和分支比得到了令人满意的重现,从而证明了该方法的可行性。对于低对称性结构,基于组态相互作用的计算更加复杂,结果的可靠性也会降低,而从头计算多重态方法具有可以在没有任何可调参数的情况下处理任意对称性的优点。这一方法主要通过在成熟的基于单电子近似的第一性原理计算软件中添加功能扩展包来实现,例如Ikeno等的工作就是以维也纳大学开发并维护的VASP(https://www.vasp.at/)作为基础开展的。
当X射线与物质中的电子相互作用时,在特定的能量下X射线会被强烈吸收,这一能量被称为吸收边。吸收边的能量由芯电子的结合能决定,所以sXAS具有元素分辨能力。对于sXAS而言,其跃迁过程满足偶极选择规则,决定了从某个初始态开始可以达到哪个最终态以及达到什么强度,因此sXAS对初始状态的电子结构非常敏感,包括影响电子结构的自旋、轨道和价态,局域对称性等。对于sXAS光谱的分析,目前主要有两种方法,其一可以通过与已知材料光谱的对比,其二可以通过理论谱学计算的方法。因为sXAS的多重态结构能够反映材料的局域电子结构,所以可以通过谱学计算的方法定量获取体系中TM离子的精细电子结构信息,提升人们对材料微观机制的理解。
2.1 价态分析
对于同种元素的不同价态,sXAS的能量位置和形状都会有明显的差异。其中,sXAS的能量位置的差异主要来自于X射线吸收过程的末态效应。
由于库仑相互作用,金属离子的价态增加1时,sXAS的L2,3边会向更高能量移动1 eV甚至更多[28-29]。3dn和3dn-1组态之间的能量差为:
ΔE近似等于Upd与Udd的能量差(1~2 eV),其中:Udd是3d电子之间的库仑排斥能,Upd是3d电子和2p芯空穴之间的库仑排斥能。如图3(a)所示,在BaCoO3(Co4+)、EuCoO3(Co3+)和CoO(Co2+)的Co L2,3边sXAS光谱中[30],随着Co离子价态的升高,白线峰向高能方向移动。同样在SrMnO3(Mn4+)、LaMnO3(Mn3+)、MnO(Mn2+)的Mn L2,3边光谱中[31],随着Mn离子价态的升高,白线峰也向高能方向移动,如图3(b)所示。此外,谱形的不同主要来源于电子排布的差异。虽然对于非单一价态体系,利用标样谱线性叠加的方式可以实现复杂价态成分的分析。但是在某些情况下实验谱存在展宽、能量分辨率、对称性的差异等会使得我们难以获得合适的标样谱。在谱学计算中,可以通过改变初末态、展宽、对称性等方法得到各个价态的理论谱图,能够很好地解决复杂价态体系的分析。

图3 (a) 不同价态的Co L2,3边光谱:从上至下分别为BaCoO3(Co4+),EuCoO3(Co3+),CoO(Co2+)[30];(b) 不同价态的Mn L2,3边光谱:从上至下分别为SrMnO3(Mn4+),LaMnO3(Mn3+),MnO(Mn2+)[31];(c) Ba15V12S34O3的实验L2,3边光谱以及计算的VIII、VIV、VV及其和[32];(d) Sr2Cr0.5Ni0.5OsO6的Cr-L2,3边光谱,Cr2O3,PbCrO3,Ag2Cr2O7为Cr-L2,3边参考光谱[33](彩图见网络版)Fig.3 (a) Co L2,3 edge spectra of different valence states, from top to bottom: BaCoO3(Co4+), EuCoO3(Co3+), CoO(Co2+)[20]; (b) Mn L2,3 edge spectra of different valence states, from top to bottom: SrMnO3(Mn4+), LaMnO3(Mn3+), MnO(Mn2+)[31]; (c) Experimental VL2,3 XAS spectra of Ba15V12S34O3 together with calculated VIII, VIV, VV and their sum [32]; (d) The Cr-L2,3 XAS spectra of Sr2Cr0.5Ni0.5OsO6 together with Cr2O3, PbCrO3, and Ag2Cr2O7 as references[33] (color inline)
在一个晶格中同时稳定三种氧化态是一个挑战,并且需要广泛的主晶格设计来防止相分离。为此,Wong等[32]使用sXAS并组态相互作用计算研究了新型化合物Ba15V12S34O3是否具有VIII/IV/V三种氧化态,结果表明,V3+、V4+和V5+的理论光谱可以很好地再现Ba15V12S34O3的所有实验光谱特征,证明了该化合物能稳定存在三种价态的V离子,如图3(c)所示。
Chen等[33]在高温高压条件下成功合成了具有高TC铁磁性的Sr2Cr1-xNixOsO6,其中Sr2Cr0.5Ni0.5OsO6的磁化强度是Sr2CrOsO6的6倍。为了阐明Sr2Cr0.5Ni0.5OsO6结构产生高TC铁磁性的原因,作者结合sXAS的研究表明,Sr2Cr0.5Ni0.5OsO6中Cr为2/3的Cr3+和1/3的Cr6+的混合价态构型(图3(d)),表明Ni的掺杂并未引起Os的价态变化,Os与Cr呈反铁磁耦合,而与Ni呈铁磁耦合,铁磁性的Ni取代了反铁磁性的Cr,解释了铁磁性体系中净磁性的增加。
Joy等[34-35]合成了一种具有双相结构的LaMn0.5Co0.5O3材料,并通过磁化率和X射线光电子能谱推断出LaMn0.5Co0.5O3中具有较高TC的相含有HS-Mn3+和LS-Co3+,而较低的TC相含有Co2+和Mn4+。但Burnus等[31]使用sXAS实验结合组态相互作用的计算合理地发现,LaMn0.5Co0.5O3中具有高TC的相应具有HS-Co2+和HS-Mn4+,低TC相应具有LS-Co3+,而完全不同XPS的结果,如图4(a)和(b)所示。表明与XPS实验和磁化率实验相比,sXAS是探测局部电子结构信息更准确的方法之一。
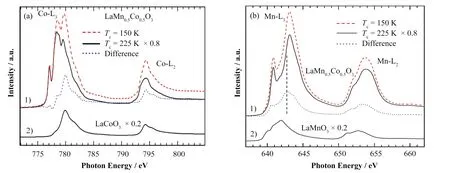
图4 (a) 1) TC = 225 K和TC = 150 K的LaMn0.5Co0.5O3样品的Co-L2,3 XAS光谱,以及它们的差分谱,2) LaCoO3作为Co3+参考[31];(b) 1) TC = 225 K和TC = 150 K的LaMn0.5Co0.5O3样品的Mn-L2,3 XAS光谱,以及它们的差分谱,2) LaMnO3作为Mn3+参考[31]Fig.4 (a) 1) Co-L2,3 XAS spectra of a the LaMn0.5Co0.5O3 samples with TC = 225 K and TC = 150 K, their difference, and 2) LaCoO3 as Co3+ reference[31]; (b) 1) Mn-L2,3 XAS spectra of a the two LaMn0.5Co0.5O3 samples with TC = 225 K, TC = 150 K, their difference ,and 2) LaMnO3 (Mn3+) for comparison[31]
2.2 对称性分析
在一个原子位置上所有其他电荷的电势称为马德隆势,一般我们只需考虑马德隆势的局域值,马德隆势的导数可以用来描述局域的晶体场。晶体场可以用来反映TM离子局域对称性的电子轨道,会影响3d轨道的劈裂情况,进而对sXAS的谱形产生影响。
虽然YBaCo3AlO7[36]、CoO和Ca3CoRhO6[37]都是具有Co2+的3d7结构,但是它们的Co离子分别以四面体对称结构(Td)、八面体对称性结构(Oh)和三角对称性结构(D3d)的形式存在。对称性的不同使得它们电子填充的轨道以及轨道之间劈裂的能级差都有所不同(图5(b)),从而产生谱形的差异,如图5(a)所示。
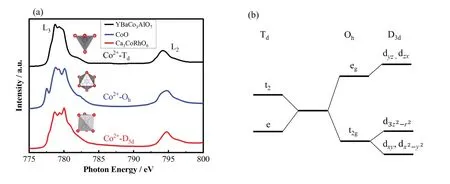
图5 (a) 不同对称性的Co L2,3边实验光谱:从上至下分别为YBaCo3AlO7(Co2+-Td)[36]、CoO(Co2+-Oh)和Ca3CoRhO6(Co2+-D3d)[37];(b) 三角对称(D3d),八面体对称(Oh),四面体对称(Td)性下的d能级劈裂Fig.5 (a) Experimental spectra of Co L2,3 edge with different symmetries: YBaCo3AlO7(Co2+-Td)[36], CoO(Co2+-Oh), and Ca3CoRhO6(Co2+-D3d)[37]; (b) Energy-level diagram for the d shell in trigonal (D3d), octahedral (Oh), and tetrahedral symmetry (Td)
Lee等[38]通过sXAS技术探究了Sr2Mn1-xMoxO6中Mn和Mo的价态随Mo掺杂浓度(x)的变化,当x从0增加到0.5时,Mn的价态逐渐降低,从x=0时的正四价变为x=0.5时的正二价。当x=0、0.3、0.5时,Mn的实验谱图分别与Oh对称的Mn4+、D4h对称的Mn3+以及Oh对称的Mn2+计算结果一致,表明在Mn3+中存在着不可忽略的姜-泰勒(J-T)效应。
2.3 自旋态分析
sXAS对TM元素的自旋态极其敏感,对于3d TM,多重态效应、晶体场强度和电荷转移能的大小等都会影响TM离子的自旋态,这将强烈影响材料的电荷输运性质和磁性等,并产生谱线的差异。不同自旋状态的稳定性取决于洪德交换能和晶场劈裂能之间的竞争。通常共价性的增强有利于低自旋构型,因为强共价性导致原子内相互作用的减弱,从而增强了晶体场强度[39-41]。在EuCoO3(LS-Co3+)和Sr2CoO3Cl(HS-Co3+)的Co L2,3边sXAS光谱中[30],Co离子均为正三价,而由于不同的自旋态会有不同的轨道占据,进而导致谱形的变化,如图6(a)所示。在谱学计算中,可以通过调整晶体场劈裂能、电荷转移能、自旋-轨道耦合作用等参数,实现对元素自旋态的分析,得出不同自旋态下金属元素电子构型的差异,及其产生不同性质的原因。
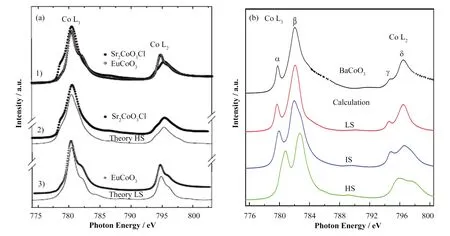
图6 (a) 1)分别为EuCoO3(LS-Co3+)和Sr2CoO3Cl(HS-Co3+)的Co L2,3边光谱,2) HS的Sr2CoO3Cl光谱与理论模拟的比较,3) LS的EuCoO3光谱与理论模拟的比较[30];(b) BaCoO3的L2,3边光谱和计算了具有3d5构型的LS、IS和HS的Co L2,3边光谱[49]Fig.6 (a) 1) The Co L2,3-edge spectra of EuCoO3(LS-Co3+) and Sr2CoO3Cl(HS-Co3+), 2) Comparison between the Sr2CoO3Cl spectrum and a theoretical simulation for high-spin (HS), 3) Comparison between the EuCoO3 spectrum and a theoretical simulation for low-spin (LS)[30]; (b) The Co L2,3-edge spectra of BaCoO3 and calculated Co L2,3 XAS spectra for a CoO6 cluster with 3d5 low-spin(LS), intermediate-spin (IS), and high-spin (HS) state configurations[49]
钴酸盐丰富的电子和磁性能不仅与钴在不同价态稳定的可能性密切相关,而且与自旋态自由度密切相关[42-46]。例如,在八面体中,分别具有形式d6和d5构型的Co3+和Co4+离子可以存在三种可能的自旋状态:HS、LS甚至IS。BaCoO3由在c轴面共享的CoO6八面体一维链组成[47-48],Chin等[49]结合sXAS和组态相互作用模型对BaCoO3的局部电子结构进行了详细的研究。结果表明,Co离子为低自旋的四价3d5态,如图6(b)所示。
在3d过渡金属氧化物体系中,钴酸盐具有很高的自旋自由度。Haw等[50]探究了LaSrCoO4中Co3+的自旋态。LaSrCoO4的sXAS光谱结合组态相互作用计算的结果表明,Co3+的实验谱可以由HS-Co3+和LS-Co3+的计算谱线性叠加再现。在进行组态相互作用计算过程中也分别考虑了不同电子排布情况下的IS态Co3+,但得到的谱图与实验谱均存在差别,表明LaSrCoO4中不存在IS-Co3+。
电荷传输研究表明,A位有序钙钛矿YCu3Co4O12是一种绝缘体,而CaCu3Co4O12表现出金属行为[51]。为了深入理解引起该转变的原因,Chin等[52]对YCu3Co4O12和CaCu3Co4O12中Co的自旋态做了详细探究。sXAS实验和组态相互作用计算结果表明YCu3Co4O12中Co以LS-Co3+的形式存在,CaCu3Co4O12中Co以75% LS-Co3+和25% LS-Co4+的形式存在。在CaCu3Co4O12中,相邻的LS-Co3+和LSCo4+离子之间的电荷交换不会受到自旋阻断机制,从电子结构角度合理地解释了YCu3Co4O12为绝缘体而CaCu3Co4O12为导体。
LaMnO3是一种反铁磁绝缘体,而在Mn位置掺Co的LaMn1-xCoxO3具有铁磁性。Bhat等[53]探究LaMn1-xCoxO3体系的磁性转变。结合sXAS实验和组态相互作用的计算表明,x=0.1和x=0.2中Co是以HS-Co2+存在的,随着Co含量的增加,在x=0.3,0.4,0.5中均存HS态和LS态Co3+。在所有掺杂体系中,Mn均以Mn3+和Mn4+的混合价态形式存在,并且随着掺杂Co浓度的增加,Mn3+的比例不断增加。分析可知,Co和Mn离子不同价态之间的交换相互作用使得LaMn1-xCoxO3具有铁磁性的。
过渡金属酞菁化合物(Transition Metal Phthalocyanines,TMPcs)的基态可以具有多种自旋态,其自旋态决定了本身独特的光学、磁学等性质[54],目前仍是研究的一大热点。例如,Belser等[55]研究了在Cu(111)和Ag(111)上生长的含氟FePc薄膜的自旋态与薄膜厚度的关系。sXAS光谱结果表明,当薄膜厚度增加时,Fe离子的L2,3边会发生剧烈的改变。结合组态相互作用计算对Fe离子的研究表明,当薄膜厚度逐渐增加时,Fe离子的自旋态发生改变,表明不同的sXAS峰形与FePcF16分子中Fe离子的详细电子排布方式有关。
2.4 轨道极化分析
sXAS能够直接获取未占据的3d空轨道的信息,特别是线性偏振的XAS(LD-XAS)能够利用X射线吸收光谱中的偏振依赖性直接确定空穴所占据的轨道。利用组态相互作用计算,通过理论谱和实验谱之间的拟合,进而可以由空轨道的信息推演出占据轨道的信息。在进行谱学计算时,可以通过构造多组初末态、调整参数等方法来分析TM离子具体的轨道劈裂和轨道占据形式,进而分析由电子占据改变引起的特性变化。此外,使用sXAS方法研究Heusler等合金体系的铁磁等特性也是一个重要方向。遗憾的是,本文主要涉及的配位场理论并不适用于金属键的计算。到目前为止,还没有可靠的理论能够实现金属体系的sXAS计算,这还是一块待开发的领域。
Haw等[56]采用脉冲激光沉积法在YAlO3(010)衬底上制备了b轴定向正交的YMnO3(o-YMnO3)外延薄膜,研究了o-YMnO3中的轨道占据和姜-泰勒畸变。结合sXAS实验与组态相互作用计算,分析了Mn离子的3d电子占据态和eg态的姜-泰勒劈裂信息。结果表明,Mn离子的eg的电子显示出强的轨道极化(图7(a)),在ab平面中具有d3x2-r2类型的交错轨道排列方式,其抑制了最近邻间的交换相互作用,增强了次近邻间的超交换相互作用。
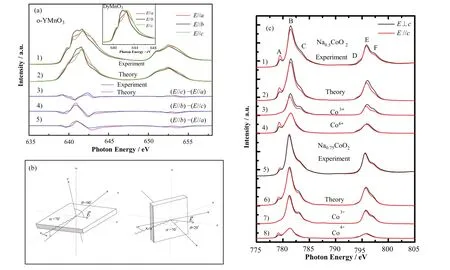
图7 (a) 1) YAlO3(001)衬底上o-YMnO3的实验和2)理论极化相关的Mn L2,3边XAS,实验和理论LD光谱,3) (E//c)-(E//a),4) (E//b)-(E//c)和5) (E//b)-(E//a),附图为o-DyMnO3晶体偏振依赖的Mn L3边[56];(b) LD-sXAS的光路示意图,θ为电场矢量与c轴表面法线之间的夹角,α为坡印亭矢量与表面法线之间的夹角[57];(c) 偏振依赖的Na0.5CoO2和Na0.75CoO2的Co L2,3边光谱,其中1)和5)为实验光谱,3)和7)为Co3+的理论谱,4)和8)为Co4+的理论谱,2)和6)为相应的拟合理论谱[57]Fig.7 (a) 1) Experimental and 2) theoretical polarization-dependent Mn L2,3-edge X-ray absorption spectra of o-YMnO3 on a YAlO3(001) substrate, experimental and theoretical LD spectra of 3) (E//c)-(E//a), 4) (E//b)-(E//c), and 5) (E//b)-(E//a). The inset shows the polarization-dependent Mn L3-edge absorption spectra of the o-DyMnO3 crystal[56]; (b) Experimental geometry with polarization of the light. θ is the angle between the electric field vector and the c-axis surface normal, and α is the tilt between the Poynting vectorand the surface normal[57]; (c) Polarization-dependent Co L2,3 XAS spectra of Na0.5CoO2 and Na0.75CoO2, where 1) and 5) are the experimental spectra; 3) and 7) are the theoretical spectra of Co3+, 4) and 8) are the theoretical spectra of Co4+, and 2) and 6) are the corresponding fitted theoretical spectra[57]
Lin等[57]对NaxCoO2中的Co的局域电子结构,尤其是轨道极化等电子排布方式进行了研究。NaxCoO2是CoO2与Na沿c轴交替排列而成,其中八面体CoO6沿着c轴有明显的压缩,因此,中心离子Co具有D3d对称性。在LD-XAS实验时,样品相对于入射光束是倾斜的,因此光的坡印亭矢量与表面法线成70°角。改变偏振方向时,样品可以绕着坡印亭矢量的轴进行旋转,如图7(b)所示。电场矢量E→和c轴之间的角度θ可以在20°~90°之间变化。这种方法允许入射光束的光路独立于θ,保证了作为偏振函数的谱线形状的可靠。结合组态相互作用的计算,分别对沿着垂直于c轴与平行于c轴的LD-sXAS的Co-L2,3边光谱进行分析,如图7(c)所示。发现Na0.5CoO2比Na0.75CoO2的偏振特征越明显,表明电子在Na0.5CoO2轨道上的占据具有很强的各向异性。Pellegrin等[58]在分析La2-xSrxCuO4体系时,采用偏振相关X射线吸收光谱以位置选择方式测量局域未占据态以及对这些态有贡献的轨道对称性。结果表明,Sr掺杂后电子主要占据在面内的O-2px,y轨道和Cu-3dx2-y2轨道。在几乎所有系统中,在较高的能量下,观察到具有Cu 3d3z2-r2特征的高光谱权重,这可能是Cu-4s和Cu-4p态的杂化引起的。
2.5 电荷转移分析
3d TM离子和周围配体之间的成键作用能明显地影响sXAS光谱的线形。特别对于高价态的过渡金属,例如Co4+、Ni3+等,中心离子和配体之间的电荷转移作用明显,具有配体空穴的组态在初态波函数中占据主要成分,会导致sXAS光谱中弱卫星峰的形成和多重态结构的收缩。因为电子和轨道之间的相互作用复杂,所以难以直接从实验谱中得到成键与电荷转移的相关信息。利用谱学计算,通过多组态线性叠加并考虑配体与TM离子之间的电荷转移,可以很好地再现sXAS光谱中弱卫星峰和多重态结构的收缩现象,进而从中提取出TM离子和周围配体之间的成键强度、电荷转移程度等电子结构信息。
Mizokawa等[59]使用sXAS并结合组态相互作用模型对NaCuO2的局域电子结构进行了研究。结果表明,其电荷转移能为负,基态以d9构型为主,强3d与配体之间的杂化打开了负电荷转移能的带隙,如图8所示[60]。该带隙对应的电荷波动主要为p-p型,而不是传统的莫特-哈伯德(d-d)型。间隙的大小受金属氧局部单元的几何排列的强烈影响。
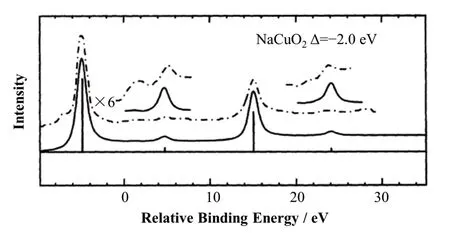
图8 NaCuO2中Cu L2,3边的计算谱图(实线)[59]与实验谱图(虚线)[60]的比较Fig.8 The comparison of theoretical spectra (solid curve)[59]and experimental spectra (dash-dotted curve)[60] about Cu L2,3-edge in NaCuO2
在TM基有机化合物中,温度、压力、光辐射和电场等都会引起TM和C/N之间复杂的电荷转移作用产生电子结构的变化[61-63],因此具有独特的物理化学性质而备受关注。例如,Zhou等[64]利用谱学计算研究了CoPc分子体系中Co2+的3d基态构型。结合组态相互作用计算结果表明,基态由73% 3d7和27% 3d8的Co离子组成。采用各向同性的成键作用模型难以很好地再现实验数据,说明离子与配体之间的相互作用存在较强的各向异性。Nanba等[65]指出对于Fe(CN)6体系,如果认为不存在金属与配体之间的电荷转移或只存在金属到配体的电荷转移,拟合得到的谱图与实验谱相差较大。当计算时考虑金属与配体之间存在较强的电荷转移,且为金属3d轨道到配体的电荷转移时,得到的谱图与实验谱吻合较好。
3 谱学计算在分析储能电池和电催化等电化学机制中的应用
结合谱学计算技术,sXAS能够详细分析电子(或空穴)在组成离子上的分布,并获取它们的价态、自旋态和晶体场等信息,对于理解3d TM基化合物特殊的物理性质十分重要,在物理学领域取得了巨大成功。目前这一方法也引起了电化学领域学者的兴趣。电化学反应过程的本质特点在于反应过程中会发生电子转移,因此获取反应过程中体系的电子结构信息对人们理解电化学反应机制有着重要的意义。sXAS技术,特别是原位sXAS实验技术的发展,使得人们能够更好地理解反应过程中的电子结构的演变过程[66]。电子结构信息的精确提取不仅依赖于sXAS实验手段的发展,同样也依赖于相应的谱学理论计算方法的发展和应用。
3.1 电池领域的应用
随着全球能源危机的加剧,越来越多的人意识到了清洁能源的重要性。新能源电池的潜在应用也吸引着众多科研人员的关注。由于TM离子具有丰富的物理化学特性,尤其是3d过渡金属逐渐体现出在电极材料中的优越性,sXAS可以用来研究电池电极TM离子的氧化态、化学键、自旋态和轨道特性等,在电池电极的发展过程中起到了重要作用。
尖晶石结构的LiNi0.5Mn1.5O4因具有对环境友好、成本低以及高能量密度的特点而备受关注。其较高的能量密度源于4.7 V的高工作电压,主要由Ni2+↔Ni4+的氧化还原反应决定,而Ni2+↔Ni4+的反应是双电子转移过程还是单电子转移过程仍然不清楚,因此理解该氧化还原相关的电子转移过程对于理解其电化学性能至关重要。为了深入探究LiNi0.5Mn1.5O4在充放电过程中的电子转移机制,Qiao等[67]对其进行了sXAS实验,并采用安德森杂质模型对八面体对称的Ni2+、Ni3+和Ni4+进行了计算,通过对比实验谱可知,未充电阶段、充电50%阶段和完全充电阶段分别对应于Ni2+、Ni3+和Ni4+,如图9(a)所示。表明Ni3+是LiNi0.5Mn1.5O4电极在电化学反应中形成的稳定中间态,也就是说LiNi0.5Mn1.5O4电极在充放电过程中发生了两个单电子的转移过程,即Ni2+↔Ni3+和Ni3+↔Ni4+,并且两个氧化还原反应发生的电位非常接近。
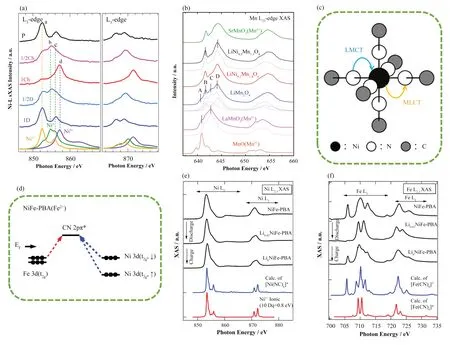
图9 (a) 充放电到不同状态的LiNi0.5Mn1.5O4的Ni L3,2边实验谱和Ni2+,Ni3+,Ni4+的计算谱[67];(b) LiMn2O4、LiNi0.2Mn1.8O4、LiNi0.5Mn1.5O4的L2,3边XAS光谱,以及作为标样的MnO(Mn2+),LaMnO3(Mn3+),SrMnO3(Mn4+)的光谱,拟合的LiMn2O4和LiNi0.5Mn1.5O4的L2,3边XAS光谱也显示在相应的实验谱下面[75];(c) MLCT和LMCT模型[81];(d) NiFe-PBA从Ni 3d(t2g)轨道到反键轨道的电荷转移[81];(e) LixNiFe-PBA的Ni L2,3边光谱,[NiII(NC)6]4-团簇的理论谱和Ni2+离子的理论谱,对谱线进行了洛伦兹和高斯展宽(每个宽度为0.25 eV)[81];(f) LixNiFe-PBA的Fe L2,3边光谱,[FeIII(CN)6]3-团簇和[FeII(CN)6]4-团簇的理论谱[81](彩图见网络版)Fig.9 (a) Ni L3,2 edge XAS spectra of LiNi0.5Mn1.5O4 in different states and the calculated spectra of Ni2+, Ni3+, and Ni4+[67]; (b)Experimental Mn L2,3 XAS spectra of LiMn2O4, LiNi0.2Mn1.8O4, and LiNi0.5Mn1.5O4, as well as the relevant spectra of MnO(Mn2+),LaMnO3(Mn3+), and SrMnO3(Mn4+, green). The simulated Mn L2,3 XAS spectra of LiMn2O4 and LiNi0.5Mn1.5O4 are also shown below the corresponding experimental spectra[75]; (c) MLCT and LMCT models[81]; (d) CTs from Ni 3d(t2g) orbital to antibonding-type for NiFe-PBA[81]; (e) Ni L2,3 edge XAS spectra of LixNiFe-PBA; the theoretical spectrum of the [NiII(NC)6]4- cluster for NiFe-PBA and that of the ionic Ni2+ state are also shown. The theoretical spectra were convoluted from the line spectra, using Lorentzian and Gaussian functions (with each width 0.25 eV)[81]; (f) Fe L2,3 edge XAS spectra of LixNiFe-PBA, and the theoretical spectra of the[FeIII(CN)6]3- and [FeII(CN)6]4- clusters for NiFe-PBA[81] (color online)
磷酸铁锂(LiFePO4)由于廉价的组成元素和具有高热力学稳定性,因此在锂离子电池领域得到了广泛的研究[68-69]。然而,其氧化还原电位相对于Li/Li+约为3.45V,明显低于其他阴极材料,限制了其应用,如LiCoO2和LiMn2O4等[70]。为了提高氧化还原电位,Asakura等[71]用Mn部分代替Fe,对LiMn0.6Fe0.4PO4纳米线的放电过程进行了研究。Mn的sXAS实验谱线特征在放电开始和结束阶段基本保持不变,只是在放电结束阶段,在L3边前出现一个小的肩峰。利用组态相互作用计算分析可知体系中Mn在放电开始和结束阶段价态与自旋态始终保持为HS-Mn2+,肩峰来源于不同方向键长的改变所引起的晶体场劈裂能的增加。而Fe的sXAS实验和计算结果表明在整个放电过程中Fe被大部分还原为Fe2+,仅有小部分未改变价态。结合O K边的变化,得知O 2p轨道与Fe 3d轨道杂化对整个充放电过程中的氧化还原反应至关重要。特别是充放电曲线中的4.0 V平台应归因于Fe 3d轨道与O 2p轨道之间的电荷转移。
在锂离子电池阴极材料体系中,除了金属磷酸锂体系优异的性能外还有Li2MnO3、LiCoO2、LiNiO2等层状材料固溶体因其高容量和良好的循环寿命而备受关注[72]。其中,Li2MnO3材料的容量高达300 mAh/g,几乎是目前广泛使用的LiCoO2等阴极材料的两倍[73]。先前对Li2MnO3充放电过程的研究表明,存在Mn氧化还原(Mn4+↔Mn5+)机制的可能。为了验证这一观点,Kubobuchi等[74]使用sXAS及从头算多重态计算方法对Li2MnO3充放电过程的机理进行了探究。研究结果发现,在充放电过程中,Mn的sXAS实验谱具有相似的形状,只是峰的强度存在细微差别。这表明Mn的价态在整个过程中没有发生变化,并且在Mn4+假设的计算下基于Mn4+的理论计算可以很好再现实验谱。计算结果显示,随着Li组分的减少,L3边前峰的强度有所减小,而L3边主峰的强度略微增强,表明其峰强度取决于Li的组成,光谱的变化不是由于Mn的价态而引起的,而是由于Li离子脱嵌过程中Mn离子周围原子配位的变化而引起的。
锂离子电池充放电过程中TM离子和配体离子之间的电荷转移作用对其稳定性和性能都有着重要影响。Liu等[75]探究了LiNixMn2-xO4阴极材料在充放电过程中TM离子的价态变化及其与O配体之间的电荷转移情况。XAS结果表明,随着Ni的掺入,体系中的Mn4+含量不断升高,组态相互作用计算结果表明,Mn4+的基态由23%的3d3、56%的3d4和21%的组成,Mn3+的基态由57%的3d4、39%的和4%的组成(图9(b)),因此体系中Mn-O键具有高度共价性。此外配体空穴组分在Mn4+的基态中占主导地位,当Mn离子的价态增加1时,50%的电荷变化是由O 2p提供的,表明了TM和O之间的电荷转移对电化学性能的重要作用。
普鲁士蓝类似物具有独特的电子特性,成功应用于高功率、低成本和耐用的可充电电池[76-79]。Firouzi等[80]研究了一种钠离子电池的阳极材料六氰基锰酸锰。进行的sXAS实验和组态相互作用计算结果,首次证实了阳极存在Mn+,充放电过程中氧化还原反应为(Mn2+↔Mn+),结果表明,Mn 3d与配体CN之间存在强的杂化作用,这有利于提高电池性能。Nanba等[81]对NiFe-PBA在锂离子嵌入和脱出过程进行了sXAS实验并采用组态相互作用计算对氰化物电极的电子结构进行了详细探究。由于CN配体具有施主与受主能力,PBA电极存在很强的金属与配体之间的双向电荷转移。计算中,考虑中心原子与配体之间的相互作用时分别采取了金属到配体的电荷转移模型(Metal-to-Ligand Charge Transfer,MLCT)与配体到金属的电荷转移模型(Ligand-to-Metal Charge Transfer,LMCT),如图9(c)所示。结合组态相互作用计算发现,无论锂离子浓度如何变化,Ni都保持为HS-Ni2+,而锂离子的脱嵌过程存在(LS-Fe3+↔LS-Fe2+)的氧化还原,如图9(e)和(f)所示。计算结果揭示了MLCT和LMCT对Fe-C和Ni-N键的贡献,其中MLCT主导了Ni-N和Fe2+-C键的电子构型(如图9(d)右图所示)。而MLCT和LMCT对Fe3+-C键的电子构型都具有重要的作用。同样在考虑TM与CN之间的双向电荷转移作用的基础上,Asakura等[82]使用sXAS研究了MnFe-PBA在可充电锂离子储能反应过程中的电子结构变化。Mn的L2,3边的结果表明,充放电过程中锂离子浓度不会改变Mn的价态和自旋态,一直保持为HS-Mn2+,CTM4XAS计算表明,Mn离子周围的晶体场作用较弱。而Fe的sXAS光谱揭示了在充放电过程中,伴随着锂离子的嵌入和脱出发生了Fe3+与Fe2+的氧化还原转变。对Fe进行的CTM4XAS计算和实验谱的对比表明,Fe和CN之间同时存在配体到金属和金属到配体之间的电荷转移,是其稳定的锂离子储存性能的主要来源。
由于电池的充放电状态能够不依赖于电压而稳定存在,通过真空转移腔将样品直接从氩气保护的手套箱转移到sXAS测试真空腔中,有效地避免水氧环境对样品的破坏,使得软X射线谱学技术能够直接应用于充放电过程中电子结构演变的探测中,极大地促进了这一技术在二次电池领域的应用。近年来随着对电池充放电的动力学过程的研究兴趣增加,人们也进一步开展了电池材料的原位sXAS研究。软X射线极短的穿透深度要求实验必须在超高真空的环境中进行,极大地增加了原位实验的难度。虽然人们在Mg离子电池电解质(主要利用Mg Kedge)[83-84]、金属空气电池(主要利用O K-edge)[85]和电极材料热稳定性变温原位sXAS实验[86]等方面取得了一些成功,但是由于完整的离子电池的结构复杂性,对于真实离子电池体系的原位sXAS实验报告仍然极少。Liu[87]等通过在电池制备过程中优化集流体和采用聚合物电解质(图10(a)和(b)),利用原位sXAS实验技术研究比较Li(Co1/3Ni1/3Mn1/3)O2(NMC)和LiFePO4(LFP)两种类型锂离子电池的电子动力学过程。研究结果表明,NMC电极能迅速响应不同的充放电状态(光谱发生变化),并定量跟踪整体荷电状态,表明电极上的荷电状态分布均匀。相反,LFP电极存在较强的弛豫效应(光谱几乎不变)和荷电状态梯度分布效应,如图10(c)和(d)所示。这项对比研究证明了原位sXAS在揭示电池电极充放电动力学方面的优势。
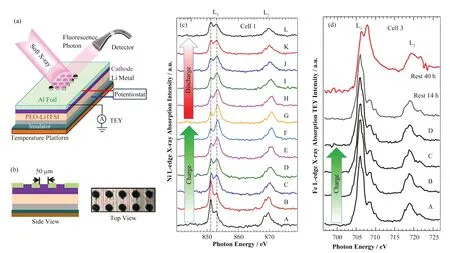
图10 (a、b) 用于sXAS原位池实验装置的示意图,用高精度激光在集流器上钻出直径为50 mm的孔阵列,入射的软X射线束和激发的荧光光子穿过集电器上的孔阵列[87];(c) NMC阴极的Ni L2,3边sXAS(TFY模式),在不同充放电状态取的光谱用A-L表示[87];(d) LFP的原位Fe L2,3边sXAS[87]Fig.10 (a, b) The schematic depiction of the experimental setup of the in situ cell for simultaneous cycling and X-ray spectroscopic measurement. An array of holes 50 mm in diameter is drilled into the current collector with a high-precision laser. The incident soft X-ray beam and excited fluorescence photon pass through the array of holes on the current collector[87]; (c) Ni L-edge sXAS TFY spectra of NMC cathode Spectra are taken at different state-of-charge levels, denoted as A-L[87]; (d) In situ Fe L2,3-edge sXAS of LFP[87]
3.2 电催化领域的应用
电催化材料在燃料电池、电解水制氢、电化学固氮和二氧化碳还原等清洁能源转化技术的发展中起着核心作用。电催化反应是一类表面反应,表面结构的变化过程对于理解反应机理至关重要。sXAS的探测深度较浅,可以用于获得材料表面的精细电子结构,如价态、电子排布、成键等信息,因此非常适合研究电催化反应。随着原位吸收谱技术的发展,人们可以动态捕获反应过程中活性位点金属的价态、自旋态、成键等的演变过程,并结合谱学计算从中提取精细电子结构信息,有助于深入理解电催化反应过程的微观机理,有效促进电催化技术的发展。
氧还原反应(Oxygen Reductive Reaction,ORR)是燃料电池中重要的半反应。ORR过程会强烈腐蚀电极,所以需要能够抵抗电化学溶解的电极材料。铂(Pt)基合金纳米颗粒在ORR过程中其他金属会溶出而形成富Pt的壳层,从而提高电催化剂的活性和稳定性。Jeon等[88]探究了Pt-Ni纳米催化剂ORR过程中Pt表面Ni的选择性溶出行为。未经处理(P)和经对苯二酚处理(HQ)的Pt-Ni的实验谱特征相似,且与理论Ni2+的谱图有相似的特征,表明两者Ni离子都被高度氧化具有相同的价态。与之相比,硫酸处理(SA)的样品L2,3边相对Ni2+的谱图向低能方向移动,表明硫酸处理使得大部分氧化镍溶解。与传统的SA处理优先失去NiO不同,HQ处理溶解了靠近表面Pt的Ni原子,使得Pt-Ni纳米颗粒具有特殊的表面组成,进而表现出优异的活性和稳定性。
钙钛矿结构的LaMnO3在碱性介质中也表现出优异的ORR催化活性[89],被认为有望替代贵金属催化剂。Ignatans等[90]探究了表面重构对LaMnO3的ORR活性的影响。sXAS实验结合组态相互作用计算分析了体相和表面Mn的价态、对称性及电子占据。在相体中,扭曲八面体产生的J-T畸变使得eg轨道劈裂,增强了Mn和O之间的电荷转移。在表面处,Mn价态相比体相略低,表面上La位点的部分缺陷会引起表面重构,使得表面层Mn2+和Mn3+的共存,这种在表面局部的TM双价态结构可能促进了ORR活性。
析氧反应(Oxygen Evolution Reaction,OER)是许多可再生能源储存和转化过程中重要的半反应,例如电解水、二氧化碳还原和金属空气电池等。钴基氧化物表现出良好的OER催化活性,为了深入理解其活性来源,Bergmann等[91]探究了八面体对称CoO、四面体对称CoO、尖晶石结构Co3O4和层状CoOOH结构中不同配位环境、价态、对称性在催化过程中活性物种的演变和活性差异。进行的sXAS和相应的从头算多重态计算结果表明,OER反应后,几种物质的Co氧化物具有相似的价态,O-2p和Co-3d轨道之间的杂化也相似。证明了钴基催化剂在反应时转化为类似的结构,而不受其初始配位环境和氧化状态的影响。
在大电流密度下开发有效的电催化剂以满足工业水分解的需求是当今的一大挑战[92]。Hu[93]等报道了一种具有五配位Co3+的BiCoO3。其在402 mV的低过电位下表现出1000 mA·cm2的大电流密度。结合sXAS光谱及其理论计算表明,BiCoO3从反应前的HS-Co3+转变为反应后的LS-Co3+的原因主要来自于OER期间由中间物种吸附过程诱导的Co3+周围的氧配位的增加。不同于人工产生的氧空位以优化中间体的结合强度来增强OER活性,BiCoO3具有丰富的活性位点和最短的反应路径,因而具有大的电流密度。
Huang等[94]在高压氧气氛围下成功制备了一种层状LiNiO2材料。结合sXAS和组态相互作用的计算表明,该材料为具有3d8构型的Ni3+结构(图11(a)),其在OER反应过程中,会发生(Ni3+→Ni4+)的价态转变,形成不寻常的双空穴结构,如图11(b)所示。这种不寻常的双空穴结构有利于晶格氧参到OER反应步骤,有效减小了O-O耦合的反应能垒,进而表现出超高的OER活性。

图11 (a) LiNiO2的Ni L2,3边XAS,以及使用3d8L构型模拟计算的光谱[94];(b) OER后LiNiO2的Ni L2,3 XAS光谱,以及75% 3d8L2和25% 3d8L构型的计算光谱之和[94];(c) CF-PBA-400的Co L2,3边拟合。拟合光谱为HS Co2+和LS Co3+的叠加,实验光谱为黑色[95];(d) CF-PBA-400的Fe L2,3边拟合。拟合光谱为LS Fe2+、HS Fe2+和HS Fe3+的叠加,实验光谱为黑色曲线[95](彩图见网络版)Fig.11 (a) Ni L2,3 XAS of LiNiO2, along with a simulated spectrum of nominal 3d8L configurations[94]; (b) Ni L2,3 XAS of LiNiO2 after OER, along with the sum (black curve) of calculated spectra of 75% 3d8L2 and 25% 3d8L configurations[94]; (c)Simulation of Co L2,3-edge of CF-PBA-400. The simulation spectrum is the sum of HS Co2+ and LS Co3+, and the experimental spectrum is shown in black[95]; (d) Simulation of Fe L2,3-edge of CF-PBA-400. The simulation spectrum is the sum of LS Fe2+, HS Fe2+, and HS Fe3+. The experimental spectrum is shown in black[95] (color online)
金属有机框架(Metal Organic Frameworks,MOFs)被认为是一类有前途的OER催化剂,因为其可调多孔结构具有较大的表面积,具有丰富的金属活性位点等。Zhou等[95]对含Co、Fe的PBA(CFPBA)进行的OER研究中结合组态相互作用的计算分析了Co离子和Fe离子的sXAS光谱。采用CoC6模型计算的LS-Co3+和HS-Co2+的组合光谱可以很好地再现CF-PBA-400的实验谱,如图11(c)所示,表明了催化剂中Co存在LS-Co3+和HS-Co2+两种形式。其中,LS-Co3+的实际价态应为+2.7,表明有0.3个电子转移到氰化物轨道。采用FeN6模型计算结果表明,Fe的光谱变化可以归因于HS-Fe3+的出现,并表明一些Fe离子不再是[Fe(CN)6]4-(图11(d)),而是在高温下发生配位结构的改变,从初始的FeC6和CoN6改变为CoC6和FeN6在电化学反应过程中确定催化剂的电子结构对于识别活性位点和反应机理非常重要。Zhou等[96]结合原位sXAS技术和组态相互作用计算探究了在OER下Li2Co2O4中Co的价态和自旋态变化。结果表明,Co离子从Co3+到Co4+的价态跃迁与时间和电压间都存在相关性,而自旋态保持不变,其中Co3.4+态为OER反应的最佳价态,如图12(a)和(b)为施加1.6 V电位20 min后和OER反应后的sXAS实验与理论拟合光谱。进一步结合密度泛函理论的计算表明,高度氧化的Co4+位点是高OER活性的主要原因,而不是Co3+位点或氧空位。Zheng等[97]探究了NiCoFeP氢氧化物在pH中性介质中的OER活性,并结合原位sXAS和谱学计算探明了NiCoFeP氢氧化物在低过电位条件下,Ni4+得到了快速地生成,进而促进OER活性。Peng等[98]结合原位sXAS(图12(c))探究了Cu2O在OER反应过程中的价态变化,如图12(d)所示。首次在OER反应过程中观察到以d9-L形式存在的高价Cu3+,表明此时Cu-3d和O-2p态之间的杂化以及O-2p轨道上额外出现的配体空穴有利于降低中间反应步骤的能垒,能够有效促进OER反应,进一步说明了TM离子的电荷和自旋态对于OER活性具有重要影响。Huang等[99]通过在Co3O4中引入丰富的缺陷位点,使其OER活性得到了大幅改善,为了理解活性提高的原因,他们利用原位sXAS技术探究了在OER下Co3O4的电子结构演变规律。原位sXAS结果表明,随着电压的升高,富含缺陷的Co3O4(D-Co3O4)中的Co2+首先转化为LS-Co3+,其中一部分LS-Co3+会进一步转化为LS-Co4+,但在无缺陷的Co3O4中虽然会有大部分Co2+转化为Co3+,但几乎没有Co3+转化为Co4+,说明了D-Co3O4反应过程中生成的LS-Co4+是OER真正的活性位点,图12(e)和(f)分别为D-Co3O4在开路和1.7 V电压下的原位sXAS光谱。
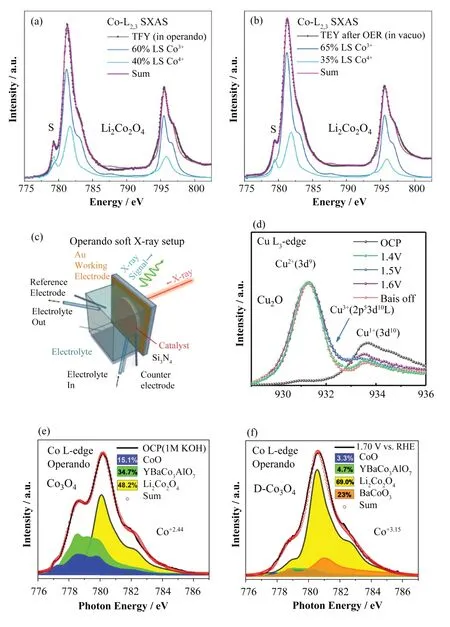
图12 (a) 在1.6 V电压下,反应20 min后,Li2Co2O4的L2,3 sXAS的实验光谱[96];(b) 在OER反应之后L2,3 sXAS的实验光谱。理论光谱由LS-Co3+光谱和LS-Co4+光谱的理论线性叠加而来[96];(c) 超高真空条件下原位软XAS装置示意图;(d) 施加不同电压时时,Cu离子在原位下的Cu L3边sXAS光谱的变化[98];(e) D-Co3O4在OCP和(f) 1.7 V vs. RHE下Co L3 sXAS光谱,光谱权重为Co2+(Oh)(CoO)、Co2+(Td)(YBaCo3AlO7)、Co3+(Oh)(Li2Co2O4)和Co4+(BaCoO3)[100](彩图见网络版)Fig.12 Experimental and theoretical spectra of Co-L2,3 sXAS spectra of Li2Co2O4. (a) Experimental Co-L2,3 sXAS spectrum of Li2Co2O4 taken in operando after 20 min under an applied voltage of 1.6 V[96]; (b) Experimental data taken in vacuo after the OER.Theoretical spectra constructed from a weighted sum of the theoretical simulation for an LS-Co3+ spectra and an LS-Co4+ spectra[96];(c) Schematic illustration of in operando soft-XAS setup under ultrahigh-vacuum condition; (d) Cu L3 edge sXAS spectra in operando at different voltages[98], Co L3 sXAS spectra of D-Co3O4 under OCP; (e, f) 1.7 V vs. RHE with spectral weights of Co2+(Oh) (CoO),Co2+(Td) (YBaCo3AlO7), Co3+(Oh) (Li2Co2O4), and Co4+(BaCoO3)[100] (color online)
4 总结与展望
本文主要介绍了sXAS理论计算的基本原理、发展现状、计算软件以及相关应用。sXAS对于TM的价态、自旋态、轨道和局域环境等电子结构信息极为敏感,结合谱学理论计算可定量获取复杂体系中TM的精细电子结构信息,从而提升人们对材料微观机制的理解,进一步指导材料结构改性推动宏观性能。在国家双碳目标的驱动下,燃料电池、电解水制氢、二氧化碳利用等电催化技术受到广泛关注,特别是低成本过渡金属基化合物催化材料被大量应用,sXAS对TM的指纹效应使其在该领域具有巨大的应用价值。
实验上,虽然sXAS可以提供丰富的电子结构信息,使人们对物理、化学、材料等领域复杂材料体系的微观机理有了深入的了解,但在未来的应用中还需要克服多种挑战。与非原位sXAS表征相比,原位sXAS技术可以通过监测真实反应条件下的电子结构变化,提供更多关于材料表面反应机制的信息。然而由于超高真空环境等因素限制,原位sXAS技术的应用仍然是一个巨大的挑战。此外,还需要设计能够产生高质量和可信实验数据的原位池。相信随着新一代具有更高通量和更高分辨率的同步辐射光源的发展,sXAS在价态、自旋态、成键等信息探测方面的独特优势将更加凸显,在发展高性能电池和电催化剂等方面发挥越来越大的作用。
理论上,基于配体场理论,人们能够实现sXAS的定量解析并从中获取诸如价态、自旋态、晶体场等电子结构信息,但是其理论本身和应用范围仍然存在一定的局限性。首先,计算时需要考虑的配体场参数复杂,包括晶体场和混合共价等因素,需要充分考虑TM离子周围的局域结构,对于复杂体系的适用性有待提高;其次,目前针对sXAS的谱学计算,大多数只考虑了中心金属离子和配体间的相互作用,忽略了中心金属离子与次近邻的金属离子之间的相互作用,解析存在一定偏差。为了解决这一问题,目前有学者基于组态相互作用模型,发展出了双团簇相互作用模型的计算方法[100],该方法允许包括成键和电荷的歧化,模型之间的库仑作用等,能够更好地再现强相互作用体系的实验谱图,但这一方法尚处于起步阶段,也没有成熟的计算软件。
在未来工作中,我们希望利用已知材料的sXAS和相应的理论计算模拟,在催化条件下对新材料进行sXAS研究,使其发挥更大的作用。
作者贡献声明樊亚磊负责论文起草、修改、搜集参考文献的主要工作;周靖、胡志伟、王建强、张林娟负责指导论文的编写,把关论文的科学性、严谨性。
