一价铜离子掺杂无铅钙钛矿Cs2AgBiBr6对晶体结构和电学性能影响第一性原理模拟研究
2024-04-29潘炀烜刘义保魏强林张子雄李凯旋
潘炀烜 刘义保,3 魏强林 张子雄 李凯旋
1(东华理工大学 核技术应用教育部工程研究中心 南昌 330013)
2(东华理工大学 核科学与工程学院 南昌 330013)
3(江西省核辐射探测及应用工程技术研究中心 南昌 330013)
卤化含铅钙钛矿材料在辐射探测领域取得了巨大的成就,但由于铅元素存在的固有毒性限制了材料工业化的应用。最近双钙钛矿A2B+B3+X6(A=Cs+,Rb+,MA+;B+=Na+,K+,Ag+;B3+=Bi3+,Sb3+;X=Cl,Br,I)由于其长久的稳定性、可调节的带隙、对环境的友好性和良好的光电性能而引起了研究人员极大的兴趣[16]。全无铅双钙钛矿Cs2AgBiBr6是目前非常有前途的光电材料,它拥有优异光电性能、良好的稳定性、较高的载流子寿命积[17],Cs2AgBiBr6单晶与多晶晶片已被用于X射线和γ射线辐射检测。目前基于无铅双钙钛矿Cs2AgBiBr6单晶制成的辐射探测器对59.5 keV γ射线的能量分辨率为13.91%[18],对比卤化含铅钙钛矿制成辐射探测器还存在不小差距。
对于双钙钛矿而言,材料的元素组成对于晶体结构稳定性与电学性能非常重要。因此可通过引入其他元素进行离子掺杂从而改变材料的晶体结构稳定性与电学性能。Jiao等[19]基于第一性原理模拟研究In3+与Sb3+掺杂对Cs2AgBiBr6的影响,模拟结构表明Cs2AgBiBr6在引入In3+或Sb3+后会材料晶格结构发生改变。Pantaler等[20]对无铅双钙钛矿Cs2AgBiBr6进行In3+掺杂实验,实验结果表明In3+会导致Cs2AgBiBr6能带从1.94 eV降为1.85 eV,掺杂产物Cs2AgBi0.25In0.75Br6为直接带隙半导体,而本征材料Cs2AgBiBr6是间接带隙半导体。Chen等[21]利用第一性原理研究了Rb+掺杂取代对无铅双钙钛矿Cs2AgBiBr6的影响,研究表明Rb+掺杂能够提高无铅双钙钛矿Cs2AgBiBr6稳定性,拓宽可见光和红外区域的光吸收。上述模拟与实验说明引入其他金属元素能够有效提高无铅双钙钛矿Cs2AgBiBr6晶体结构稳定性与电学性能。
铜是一种常见环境友好型元素,其含量丰富成本低廉。在卤化钙钛矿掺杂中Cu+应用十分广泛,较小的离子使得Cu+在对钙钛矿掺杂过程中可以灵活调节材料的晶体结构提高稳定性。De等[22]发现在热注入合成CsPbCl3的过程中,加入适量的CuCl2可以导致Cu+掺杂到纳米碳化物中,从而纠正了晶体的八面体畸变,进而提高了CsPbCl3的性能。Cu+掺杂对钙钛矿光电性能提高也具有重要影响,最近麻星宇等[23]制备了由Cu+掺杂无铅双钙钛矿Cs2AgBiBr6形成的太阳能电池,实验结果表明Cu+能够显著提高太阳能电池薄膜的质量,并且光电转换效率比原始太阳能电池提高52%。该实验说明Cu+掺杂能够提高无铅双钙钛矿Cs2AgBiBr6晶体的稳定性、光吸收能力、载流子迁移率。对于辐射探测材料而言,材料的稳定性与电学性能对能量分辨率有着重要影响。据目前已有报道而言,目前Cu+掺杂对无铅双钙钛矿Cs2AgBiBr6影响未得到系统性研究,因此本论文采用第一性原理方法计算分析Cu+掺杂无铅双钙钛矿Cs2AgBiBr6对原始材料晶体结构、电学性能影响。
1 计算方法
基于密度泛函理论(Density Functional Theory,DFT)的第一性原理,采用VASP(Vienna ab initio Simulation Package)程序[24]对Cs2Ag1-xCuxBiBr6进行晶体结构优化。其中电子与电子之间的相互作用采用广义梯度近似GGA(Generalized Gradient Approximation)下的PBE(Perdew-Burke-Ernzerhof)泛函[25]描述。电子与原子核之间的相互作用采用投影缀加平面波PAW(Projector-Augmented-Wave)赝势[26]描述。其中Cs+、Ag+、Cu+、Bi3+、Br-的价电子态分别为5s25p66s1、4d105s1、4d104s1、6s26p3、3s23p5。
在VASP的自洽计算中,平面波截断能设置为500 eV,能量与力的收敛阈值分别设置为10-5eV·atom-1和10-3eV·nm-1,高斯展宽因子设置为0.05,布里渊区设置为9×9×9的k网格点。能带计算中的布里渊区路径为W(0.000,0.250,0.750)-L(0.500,0.500,0.500)-G(0.000,0.000,0.000)-X(0.500,0.000,0.500)-W(0.500,0.250,0.750)-K(0.375,0.375,0.750)。
在能带计算中,由于PBE+SOC方法与杂化泛函HSE06方法计算量大,计算周期长,而本文关注的是Cu+掺杂对Cs2AgBiBr6电学性质影响的变化趋势,并不需要准确地计算Cs2AgBiBr6带隙值,PBE泛函方法本身能够准确地预估带隙变化趋势,而使用PBE、PBE+SOC、HSE06方法计算的能带变化趋势相似,不同的方法仅影响带隙值[27-29]。因此本文采用GGA-PBE泛函方法对能带和态密度进行计算。
2 Cu+掺杂对无铅双钙钛矿Cs2AgBiBr6结构影响
2.1 稳定性
对于双钙钛矿A2B+B3+X6钙钛矿结构的稳定性通常用两个代表性参数来估计:容差因子t和八面体因子μ[30]:
式中:rA、rB+、rB3+、rX分别是双钙钛矿中A、B+、B3+、X的离子半径。如果要形成稳定的钙钛矿结构,容差因子t需要满足0.75≤t≤1.11[31],八面体因子μ需要满足0.44≤μ≤0.90[31]。根据计算Cs2AgBiBr6、Cs2Ag0.75Cu0.25BiBr6、Cs2Ag0.25Cu0.75BiBr6、Cs2CuBiBr6如表1所示。
一是涑祊双向调水工程。在涑河上游与祊河之间开挖一条2.88 km调水水道,在调水水道两端设控制闸,连通涑河和祊河。二是引涑入青工程。在通达路桥东侧建设水闸闸室一座,通过暗涵与老引涑入青暗渠相连接,引涑河水入济青龙河水,保证临沂古护城河景观用水。三是引涑入陷工程。在涑河3号坝设置节制闸,开挖800 m的引水方涵和600 m的明渠,引涑河水入济陷泥河,补充陷泥河环境用水,改善陷泥河水质,保证陷泥河生态用水和景观用水。同时,对陷泥河、青龙河、南涑河等河道进行了拓宽、垒砌、清淤,改扩建桥涵,提高城市的排涝能力。
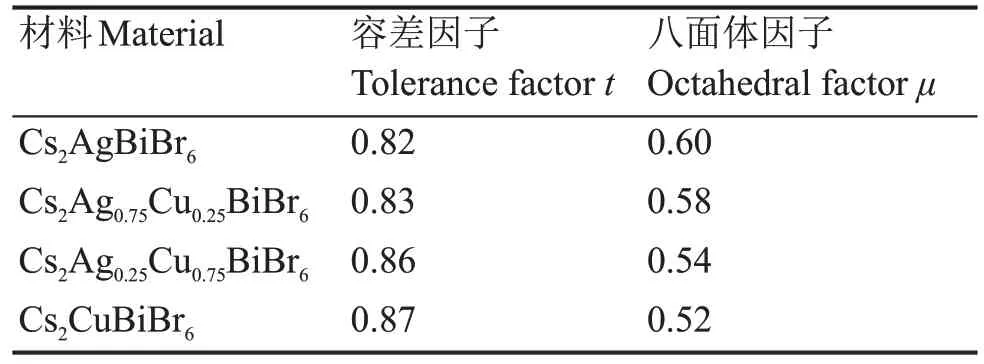
表1 无铅双钙钛矿Cs2Ag1-xCuxBiBr6的容差因子和八面体因子Table 1 Tolerance factor and octahedral factor of leadfree double perovskite Cs2Ag1-xCuxBiBr6
显然,Cs2AgBiBr6、Cs2Ag0.75Cu0.25BiBr6、Cs2Ag0.25Cu0.75BiBr6、Cs2CuBiBr6的t和μ值符合钙钛矿结构稳定性的要求,说明Cu+掺杂不会破坏Cs2AgBiBr6钙钛矿结构的稳定性。
2.2 掺杂形成能
Cs2Ag0.75Cu0.25BiBr6与Cs2Ag0.25Cu0.75BiBr6的掺杂形成能可以通过式(3~4)计算得到:
式中:ECs2Ag0.75Cu0.25BiBr6、ECs2Ag0.25Cu0.75BiBr6是指掺杂晶体Cs2Ag0.75Cu0.25BiBr6、Cs2Ag0.25Cu0.75BiBr6达到稳定结构下的最低能量;ECs2AgBiBr6是指原始晶体Cs2AgBiBr6达到稳定结构下的最低能量;μAg是指Ag的化学势能;μCu是指Cu的化学势能。经过计算,Cs2Ag0.75Cu0.25BiBr6与Cs2Ag0.25Cu0.75BiBr6的如表2所示。

表2 无铅双钙钛矿Cs2Ag1-xCuxBiBr6的掺杂形成能Table 2 Doping formation energy of lead-free double perovskite Cs2Ag1-xCuxBiBr6
形成能为负值,说明Cs2AgBiBr6在Cu+掺杂过程中的化学反应是放热过程,可以在没有外部能量的情况下自发发生。这表明Cu+掺形成Cs2Ag1-xCuxBiBr6的晶体结构比原始晶体Cs2AgBiBr6具有更高的稳定,这与麻星宇等[23]实验结果相符合,实验中Cs2AgBiBr6与Cu+掺杂的Cs2AgBiBr6作为钙钛矿型太阳能电池(Perovskite Solar Cells,PSCs)在30%的相对湿度和室温下环境下进行光电转换效率(Electron Conversion Efficiency,PCE)测试,31 d后Cs2AgBiBr6PSCs归一化的PCE从100%降为了85%,而Cu+掺杂Cs2AgBiBr6PSCs归一化的PCE从100%降为94%,也就是说Cu+掺杂可以提高Cs2AgBiBr6的稳定性。
2.3 晶格参数
Cs2AgBiBr6和Cs2Ag1-xCuxBiBr6都是具有高度对称立方晶系,其中Cs2AgBiBr6和Cs2Ag1-xCuxBiBr6优化后的结构模型如图1所示,其中Cu+掺杂取代Ag+的方式为随机取代。其中Cs2AgBiBr6结构模型如图1(a)所示,优化后的Cs2AgBiBr6超胞中包含8个Cs原子、4个Ag原子、4个Bi原子、24个Br原子。优化后的Cs2Ag0.75Cu0.25BiBr6结构模型如图1(b)所示,优化后的Cs2Ag0.75Cu0.25BiBr6超胞中包含8个Cs原子、3个Ag原子、1个Cu原子、4个Bi原子、24个Br原子。Cs2Ag0.25Cu0.75BiBr6结构模型如图1(c)所示,优化后的Cs2Ag0.25Cu0.75BiBr6超胞中包含8个Cs原子、1个Ag原子、3个Cu原子、4个Bi原子、24个Br原子。Cs2CuBiBr6结构模型如图1(d)所示,优化后的Cs2CuBiBr6超胞中包含8个Cs原子、4个Cu原子、4个Bi原子、24个Br原子。

图1 Cs2Ag1-xCuxBiBr6结构模型 (a) Cs2AgBiBr6,(b) Cs2Ag0.75Cu0.25BiBr6,(c) Cs2Ag0.25Cu0.75BiBr6,(d) Cs2AgBiBr6Fig.1 Structural model of Cs2Ag1-xCuxBiBr6 (a) Cs2AgBiBr6, (b) Cs2Ag0.75Cu0.25BiBr6, (c) Cs2Ag0.25Cu0.75BiBr6, (d) Cs2AgBiBr6
经计算优化后的晶体参数如表3所示,表中还提供了他人报道Cs2AgBiBr6与Cs2CuBiBr6晶格参数的计算值与实验值,本文计算的Cs2AgBiBr6与Cs2CuBiBr6晶格参数与他人计算的晶格参数符合良好。但与报道晶格参数实验值相比,经优化计算后的结果会略大于实验值,这是由于GGA-PBE泛函本身会高估材料的晶格参数。对比Cs2Ag1-xCuxBiBr6与Cs2AgBiBr6优化后的结构晶格参数,可知Cs2AgBiBr6在进行Cu+掺杂后会导致晶格参数减小,其原因是Ag+被半径更小的Cu+取代导致。

表3 由GGA-PBE泛函无铅双钙钛矿Cs2Ag1-xCuxBiBr6的晶格参数Table 3 Calculated lattice parameter of double lead-free perovskites Cs2Ag1-xCuxBiBr6 using GGA-PBE functional
2.4 弹性常数
材料的力学行为可以用弹性模量来解释,弹性常数决定了直接相关的弹性模量。晶体的弹性常数可以用6×6的对称矩阵来描述[35],对于立方系晶体只有三个独立的弹性常数(C11、C12和C44)。弹性常数可以用于Bardeen和Shockley[36]提出的形变势理论来预估载流子迁移率。Cs2AgBiBr6与Cs2Ag1-xCuxBiBr6弹性常数如表4所示,随着Cu+掺杂比例提高Cs2Ag1-xCuxBiBr6弹性常数会相应增加。根据形变势理论,较大的弹性常数会提供更高的载流子迁移率。

表4 由GGA-PBE泛函计算双钙钛矿Cs2Ag1-xCuxBiBr6的弹性常数Table 4 Calculated elastic constant of double perovskites Cs2Ag1-xCuxBiBr6 using GGA-PBE functional
3 Cu+掺杂对无铅双钙钛矿Cs2AgBiBr6电学性能的影响
3.1 能带分析
材料的电子状态对材料的电学性能有着重要影响,能带是表示电子状态概念的一种。能带很大程度决定了材料的电学性能。在能带结构中,处于费米能级上方的能带被称为导带,处于费米能级下方的能带称为价带。导带底部与价带顶部之间宽度被称为带隙,不同带隙可将材料分为绝缘体、半导体、导体,材料带隙的不同会导致不同的电学特性,这些特性对于在材料的技术和商业应用具有深远的影响。
为了研究Cu+掺杂对Cs2AgBiBr6电学性能的影响,本文通过PBE-GGA泛函计算了Cs2AgBiBr6与Cs2Ag1-xCuxBiBr6的能带。计算的带隙值如表5所示,由表可知,随着Cu+浓度的增加,Cs2AgBiBr6的带隙会逐渐减小,掺杂材料拥有更小的带隙值说明Cs2Ag1-xCuxBiBr6材料相比本征Cs2AgBiBr6材料具有更优的电学性能与更高载流子运输能力。Cs2AgBiBr6、Cs2Ag1-xCuxBiBr6能带如图2(a~d)所示,由能带图可以看出,Cs2AgBiBr6在引入Cu+掺杂后,会使Cs2Ag1-xCuxBiBr6导带底部显著下降而导致带隙减小。能带图中Cs2AgBiBr6与Cs2Ag1-xCuxBiBr6的导带最小值与价带最大值在K空间位置不同,可知Cs2AgBiBr6和Cs2Ag1-xCuxBiBr6都是具有间接带隙的半导体材料,Cu+离子掺杂并不会改变Cs2AgBiBr6带隙类型。Cs2AgBiBr6、Cs2Ag1-xCuxBiBr6的价带顶部略低于费米能级,说明本征材料与掺杂材料都具有P型半导体的特性。

图2 Cs2Ag1-xCuxBiBr6的能带结构 (a) Cs2AgBiBr6,(b) Cs2Ag0.75Cu0.25BiBr6,(c) Cs2Ag0.25Cu0.75BiBr6,(d) Cs2AgBiBr6Fig.2 Band structures of Cs2Ag1-xCuxBiBr6 (a) Cs2AgBiBr6, (b) Cs2Ag0.75Cu0.25BiBr6, (c) Cs2Ag0.25Cu0.75BiBr6, (d) Cs2AgBiBr6
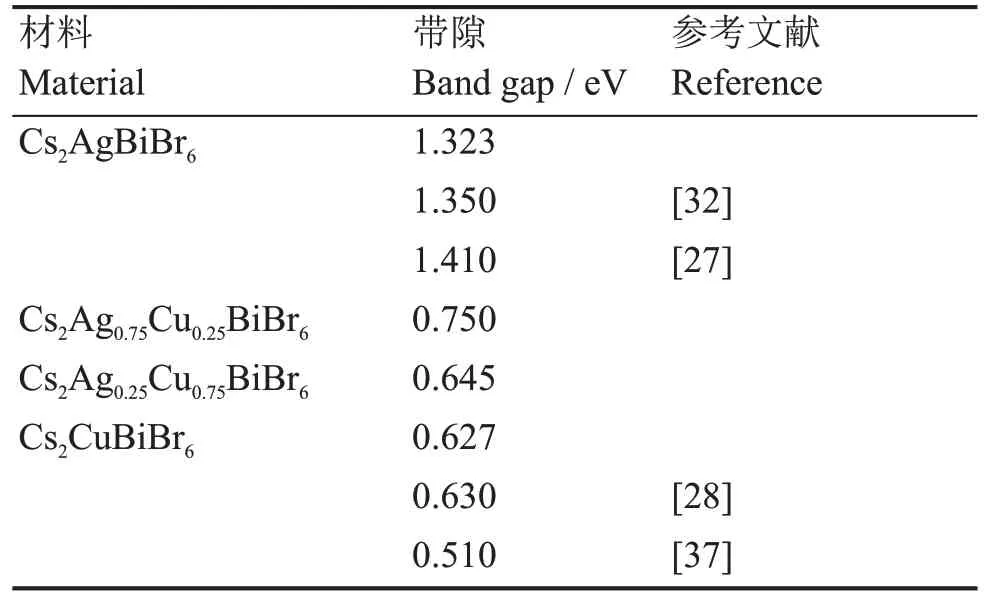
表5 由GGA-PBE泛函计算双钙钛矿Cs2Ag1-xCuxBiBr6的带隙Table 5 Calculated band gap of double perovskites Cs2Ag1-xCuxBiBr6 using GGA-PBE functional
3.2 态密度分析
为了进一步了解Cu+掺杂对Cs2AgBiBr6导带底部与价带顶部的影响,本文分别计算Cs2AgBiBr6和Cs2Ag1-xCuxBiBr6的总态密度和分波态密度。图3(a)为Cs2AgBiBr6的态密度图,从图中可知,Cs2AgBiBr6价带顶部主要由Ag4d轨道提供,导带底部主要由Bi6p轨道提供。图3(b~d)为Cs2Ag1-xCuxBiBr6的态密度图,由图可知,Cs2AgBiBr6晶体在引入Cu+离子后,原本主要由Ag4d轨道提供的价带顶部改为由Cu4d轨道提供,Ag4d轨道只对价带顶部只提供少部分贡献。带隙缩短的主要原因是Cu+掺杂比例提高导致由Bi6p轨道主要提供的导带底部逐渐靠近费米能级。
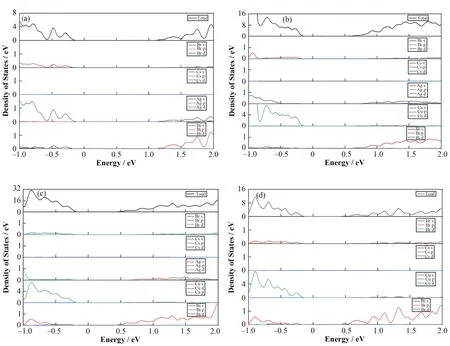
图3 Cs2Ag1-xCuxBiBr6态密度 (a) Cs2AgBiBr6,(b) Cs2Ag0.75Cu0.25BiBr6,(c) Cs2Ag0.25Cu0.75BiBr6,(d) Cs2AgBiBr6Fig.3 Densities of states of Cs2Ag1-xCuxBiBr6 (a) Cs2AgBiBr6, (b) Cs2Ag0.75Cu0.25BiBr6, (c) Cs2Ag0.25Cu0.75BiBr6, (d) Cs2AgBiBr6
4 结语
本文基于密度泛函理论通过第一性原理计算,对Cs2Ag1-xCuxBiBr6晶体结构、电学性能进行理论模拟计算。通过对比Cu+掺杂Cs2AgBiBr6前后材料结构、弹性常数、能带结构、态密度得到以下结论:
1)由于Cu+半径比Ag+小,Cu+掺杂取代Cs2AgBiBr6中的Ag+后会导致的Cs2Ag1-xCuxBiBr6晶格参数减小,但通过对Cs2Ag1-xCuxBiBr6容差因子和八面体因子计算分析可知,晶格参数的减小并不会破坏Cs2Ag1-xCuxBiBr6晶体的钙钛矿结构。Cs2Ag1-xCuxBiBr6掺杂形成能为负值,说明Cs2Ag1-xCuxBiBr6相比原始晶体材料Cs2AgBiBr6更加稳定,并且随着Cu+掺杂的比例提高,晶体的弹性常数会随之提高。根据形变势理论,随着弹性常数的提高,Cs2Ag1-xCuxBiBr6将拥有更高的载流子迁移率。
2)在Cu+掺杂Cs2AgBiBr6后形成的Cs2Ag1-xCuxBiBr6与原始材料皆是拥有间接带隙的P型半导体,并且Cs2Ag1-xCuxBiBr6相比掺杂前的Cs2AgBiBr6拥有更小的带隙值。带隙减小是由于Cs2AgBiBr6掺杂后价带顶部将由Ag4d轨道主导变为由Cu4d轨道主导,导致Bi6p轨道提供的导带底部靠近费米能级。更小的带隙值说明Cs2Ag1-xCuxBiBr6比本征材料Cs2AgBiBr6拥有更优的导电性能。
3)Cs2Ag1-xCuxBiBr6相比Cs2AgBiBr6具有更高的稳定性与更优的电学性能,Cs2Ag1-xCuxBiBr6可作为半导体辐射探测器材料。
作者贡献声明潘炀烜负责研究设计、材料仿真建模、材料性能计算、计算数据分析、文章起草与修订;刘义保负责研究设计、理论指导、审阅文章内容与修订;魏强林负责研究设计、理论指导;张子雄和李凯旋负责内容修订。
