狭缝引导配体1 3′UTR通过miR-34a-5p/SIRT1轴调节内皮细胞表型
2024-04-23胡志琴伍华燕陈凯茵徐金东方咸宏单志新
欧 涛, 胡志琴, 高 原, 伍华燕, 陈凯茵, 徐金东, 方咸宏, 单志新,*
(1)华南理工大学医学院, 广州 510006;2)广州医科大学附属第五医院药学部, 广州 510799;3)南方医科大学附属广东省人民医院(广东省医学科学院)医学研究部, 广州 510080;4)南方医科大学附属广东省人民医院(广东省医学科学院)麻醉科, 广州 510080;5)南方医科大学附属广东省人民医院(广东省医学科学院)心内科, 广州 510080)
心脏在病理条件(如心肌梗死)下会发生代偿性的心肌重构过程,其主要表现为心肌细胞体积增大,毛细血管密度降低,心肌供血不足,并可能促进心力衰竭的发生[1]。研究显示,毛细血管密度的降低一定程度上是由于血管内皮细胞功能紊乱引起,血管内皮功能损伤将导致血管新生不足、心肌缺氧、心肌收缩功能降低等一系列病理变化[2,3]。因此,研究心肌重构中血管内皮细胞功能的调节机制,对心肌重构的干预和治疗研究具有重要意义。
mRNA的3′UTR(3′ untranslated region)不但参与翻译蛋白质的过程,同时参与发挥多种生物学功能[4]。研究发现,3′UTR有一部分序列存在高度的序列保守性,含有富AU元件的3′UTR可能导致mRNA的不稳定性,促使mRNA更易降解[5]。果蝇的胚胎发育早期需要3′UTR对bicoid mRNA进行定位发挥作用,以此来产生胚胎中的蛋白质梯度[6]。在果蝇软母细胞中,Oskar mRNA其3′UTR部位能够与抑制因子相结合,抑制Oskar过早翻译,使之到达软母细胞的后极才被翻译[7]。近期研究证实,在心血管领域的研究发现,酪蛋白激酶2相互作用蛋白质-1(casein kinase-2 interacting protein-1, CKIP-1)3′UTR独立于CKIP-1蛋白发挥抑制心肌肥大作用[8]。
近年来的研究证实,狭缝引导配体(slit guidance ligand,SLIT)基因在调控血管生成中扮演着重要的角色。例如Slit 3可作为一种新型、有效的血管生成因子刺激内皮细胞增殖,可加速内皮细胞血管网络的形成,刺激体外新生血管发芽和体内新生血管生长[9]。Slit2的15外显子剪接体的编码蛋白质参与调节肿瘤周围血管正常化[10]。我们的前期工作发现,人心肌组织中SLIT1 3′UTR水平显著高于SLIT1 编码区(coding sequences, CDS),但相比于健康器官捐献者,肥厚型心肌病(hypertrophic cardiomyopathy,HCM)患者心肌中SLIT1 3′UTR水平下降,本研究旨在探究SLIT1 3′UTR对人血管内皮细胞表型的影响及可能机制,为心肌重构的干预和治疗研究提供科学资料。
1 材料与方法
1.1 心肌组织标本
利用HCM患者和健康器官捐献者的心肌组织进行SLIT1基因转录本水平的RT-qPCR检测。本文所涉及临床样本的实验均经广东省人民医院伦理委员会批准[批准号No.GDREC2019238 H(R1)],所有患者均签署知情同意书,相关心肌组织标本均由广东省心血管病研究所进行提供。
1.2 材料
主动脉内皮细胞(human aortic endothelial cells,HAECs)购自ATCC细胞库;胰蛋白酶(0.25% trypsin-EDTA)、胎牛血清(fetal bovine serum,FBS)及DMEM/F12 培养液(Gibco);细胞转染试剂Lipofectamine 2000 及Oligo 和TRIzol(Invitrogen,美国);2×pro Taq HS PCR预混液,逆转录试剂盒和2× SYBR Green Pro Taq HS Premix(湖南艾科瑞,中国);4×SDS 上样缓冲液(loading buffer)(TaKaRa,日本);miR-34a-5p mimic、mimic NC、si-SIRT1(广州锐博公司,中国);鼠抗GAPDH抗体、鼠抗SIRT1抗体(Proteintech,中国);兔抗eNOS抗体、p-eNOS抗体、VEGFA抗体(Abcam,英国);BCA蛋白质定量试剂盒、蛋白质Marker (赛默飞世尔公司,美国);PVDF膜(Whatman,英国)。ECL化学发光检测试剂盒(millipore,美国)。RIP试剂盒(广州伯信生物公司,中国)。其他涉及的生化试剂均为进口分装或国产分析纯。PCR引物由广州睿博公司合成(Table1)。
1.3 细胞培养
主动脉内皮细胞用含10%胎牛血清和100 μg/mL青霉素/链霉素的ECM培养基,在37 ℃、5% CO2的细胞培养箱中进行恒温培养,根据细胞生长状态更换新鲜培养基,当细胞密度达到90%时进行传代,取处于对数生长期且状态良好的细胞进行后续细胞腺病毒感染,mimic转染和功能检测。
1.4 重组腺病毒的制备
SLIT1 3′UTR的序列来自其mRNA的3′端非翻译区。在pAd-Track-cmv载体中的多克隆位点中定向插入SLIT1 3′UTR的模板DNA,在BJ5183大肠杆菌中与腺病毒骨架质粒pAd-Easy-I进行重组。重组成功后SLIT1 3′UTR腺病毒质粒被PacⅠ内切酶线性化,并在HEK293细胞中包装、扩增腺病毒。实验时以rAd-GFP作为对照组腺病毒(SLIT1 3′UTR和rAd-GFP的MOI均是5)。
1.5 SLIT1 3′UTR腺病毒感染与分组
0.25% EDTA胰酶消化处于对数生长期状态的细胞,以1~3×104/mL密度接种于12孔细胞培养板中,待细胞贴壁生长密度至60% ~ 70%时进行病毒感染,分为实验组和对照组,实验组和对照组分别感染SLIT1 3′UTR腺病毒和GFP空载体腺病毒,4 h时补加含10%胎牛血清的F12培养基继续培养,24 h后检测各项指标。
1.6 划痕法检测细胞增殖与迁移能力
选取处于对数生长期状态的细胞,用0.25% EDTA胰酶消化细胞,以6~9×104/mL/孔的细胞接种于6孔板中,待细胞贴壁生长密度至80% ~ 90%时进行划痕处理,倒置显微镜下观察并拍照,置于37 ℃,5% CO2细胞培养箱培养,24 h后倒置显微镜观察相同位置并拍照。
1.7 细胞成管腔能力测定
取250 μL Matrigel胶与DMEM培养基1∶1比例混合后铺于12孔板中,置于37 ℃,5% CO2细胞培养箱孵育4 h使胶凝固,每孔加入1 mL细胞数为5×106的细胞悬液,6 h后倒置显微镜下观察并拍照,对管腔结构进行量化分析。
1.8 RT-qPCR检测
利用Trizol法提取人心肌组织或者HAECs总RNA,使用分光光度计检测RNA的浓度和纯度。取2 μg总RNA进行逆转录反应后得到cDNA,通过qPCR检测SLIT1转录本、eNOS和VEGFAmRNA表达水平。另外取1 μg总RNA,加入0.2 μL 特异miRNA RT引物、5 μL 5× Prime Script RT 缓冲液、1.25 μL RT Enzyme Mix进行逆转录,得到成熟体cDNA,以其为模板进行qPCR反应,以U6作为检测内参照检测miR-27b-3p、130a-3p、34a-5p、128-3p、-7-5p表达水平。在vii A7 Quantitative PCR System(Applied Biosystems,Carlsbad,CA)进行 PCR 反应和结果分析。利用2-ΔCt或2-ΔΔCt法计算上述mRNA转录本和miRNAs的相对表达水平。本文所用PCR引物序列见Table1。

Table 1 The PCR primers sequences
1.9 Western 印迹分析
用PBS清洗细胞,弃去PBS后取适量体积的RIPA裂解液裂解细胞(加入蛋白酶抑制剂),冰上裂解细胞30 min,将裂解液转移至EP管中,于13 000×g转速下4 ℃离心10 min,收集上清液BCA法测量样品蛋白质浓度。取20 μg蛋白质,加入4×SDS上样缓冲液混匀,99 ℃变性10 min,再进行SDS-PAGE凝胶电泳,将凝胶中的蛋白质电转至PVDF膜,5%脱脂牛奶封闭1 h,分别用相应的Ⅰ抗p-eNOS(1∶1 000)、eNOS(1∶1 000)、VEGFA(1∶1 000)、GAPDH(1∶5 000)和SIRT1(1∶1 000)4 ℃孵育过夜。TBST漂洗10 min×3次,用相应的Ⅱ抗(1∶5 000)室温孵育1 h。二抗孵育完成,TBST漂洗10 min×3次,超灵敏化学发光成像仪ECL显影,使用Image J软件进行图像灰度分析,采用目的蛋白质/内参的灰度值比值比较蛋白质的表达水平,细胞内参照为GAPDH。
1.10 统计学分析
实验数据以均值±标准差( mean ±SD) 表示,采用GraphPad Prism 9. 0 软件进行统计分析。两两比较采用LSDt检验多组间均数比较先进行正态分布和方差齐性检验,方差齐性检验后采用单因素方差分析,并用Bonferroni校正的t检验进行组间两两比较。以P<0.05为差异有统计学意义。
2 结果
2.1 SLIT1 3′UTR在肥厚型心肌病人心肌中的表达
RT-qPCR检测HCM病人心肌组织SLIT1 5′UTR、SLIT1 CDS和SLIT1 3′UTR的表达情况。结果显示,与健康器官捐献者相比,HCM病人心肌中SLIT1 5′UTR水平无显著变化(Fig.1A),SLIT1 CDS水平显著升高(P<0.01, Fig.1B),而SLIT1 3′UTR水平显著降低(P<0.05, Fig.1C)。而在相对表达水平上,SLIT1 3′UTR比SLIT1 5′UTR和SLIT1 CDS水平高出1个数量级。RT-qPCR结果显示,在3种人源性的细胞中,包括人心房来源的成纤维细胞(human atrial fibroblasts, HAFs)、人主动脉内皮细胞(HAECs)和人诱导型多能干细胞来源心肌细胞(induced pluripotent stem cell derived cardiomyocytes, ips-MC),SLIT1 3′UTR表达水平显著高于SLIT1 CDS(Fig.1D)。利用糖氧剥夺实验(oxygen and glucose deprivation, OGD)处理HAECs,发现12 h和24 h的OGD处理均能有效增加HAECs中SLIT1 3′UTR水平(Fig.1E)。
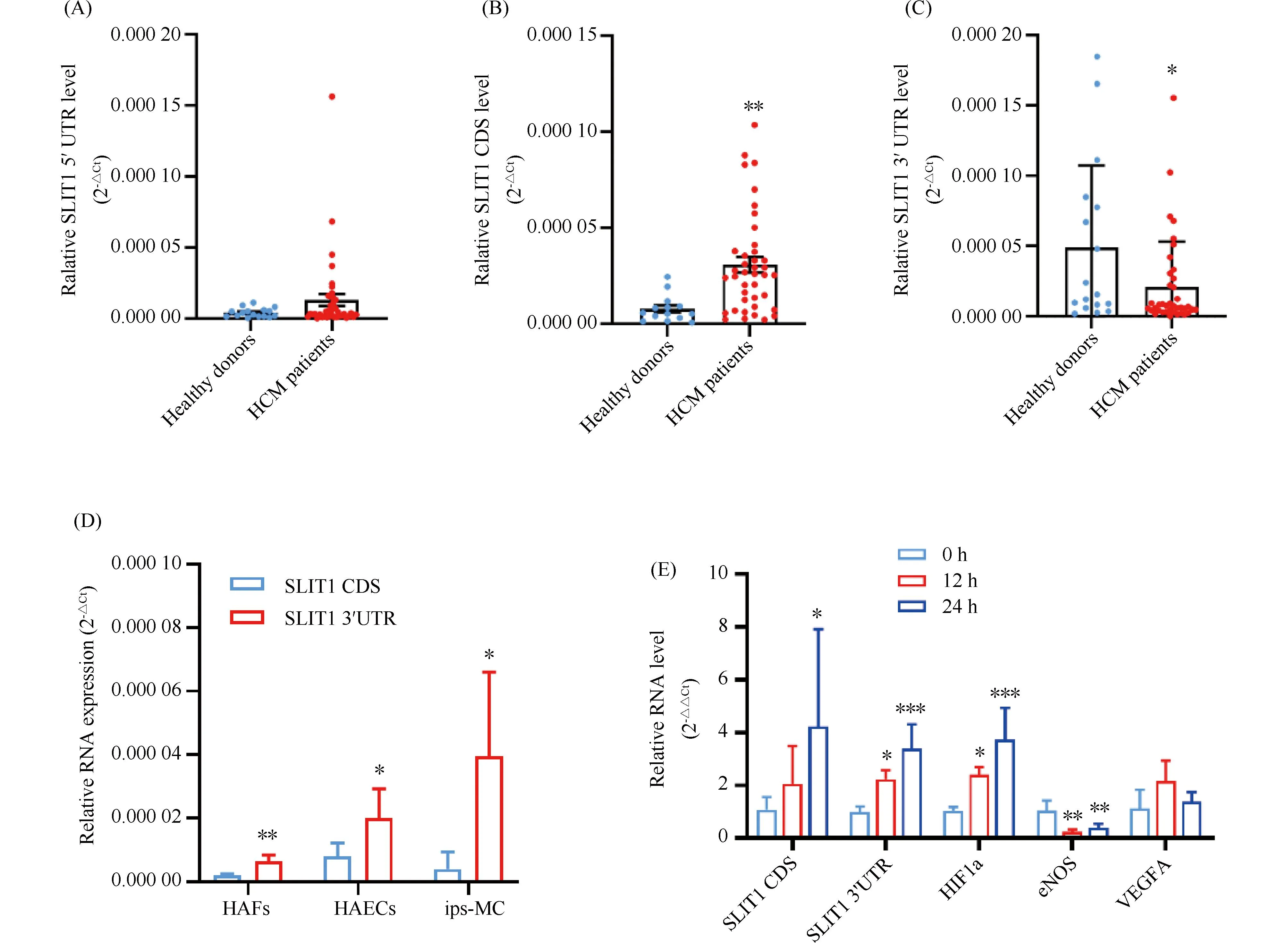
Fig.1 The level of SLIT1 3′UTR was decreased in the myocardium of HCM patients
2.2 过表达狭缝引导配体1 3′UTR促进eNOS和VEGFA表达,增加内皮细胞迁移和成管腔能力
利用SLIT1 3′UTR重组腺病毒感染HAECs,观察到HAECs有充分的绿色荧光蛋白(GFP)共表达,提示SLIT1 3′UTR在HAECs中充分表达。RT-qPCR结果证实,SLIT1 3′UTR表达水平在HAECs中显著升高(Fig.2A)。过表达SLIT1 3′UTR的HAECs中与血管生成相关的eNOS和VEGFAmRNA表达显著升高(P<0.01, Fig.2B),进一步的蛋白质免疫印迹结果显示,过表达SLIT1 3′UTR的HAECs中p-eNOS、eNOS和VEGFA蛋白质水平也一致的显著升高(P<0.01,P<0.001, Fig.2C)。
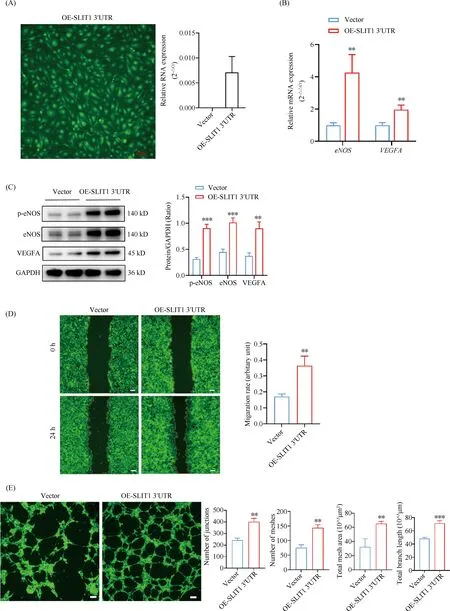
Fig.2 Overexpression of SLIT1 3′UTR promoted the migration and tube formation activities of endothelial cells
细胞划痕结果显示,过表达SLIT1 3′UTR的HAECs迁移能力显著增加(P<0.01, Fig.2D)。将腺病毒介导过表达SLIT1 3′UTR的HAECs铺于Matrigel基质胶上进行成管腔实验,结果显示,过表达SLIT1 3′UTR的HAECs体外成管腔能力显著提高(P<0.01,P<0.001,Fig.2E)。
2.3 狭缝引导配体1 3′UTR可特异结合miR-34a-5p
序列分析(http//starbase.sysu.edu.cn/starbase2/index.php)结果显示,SLIT1 3′UTR序列上有多个潜在miRNA结合位点(Fig.3A)。利用腺病毒介导在HAECs中过表达SLIT1 3′UTR,RT-qPCR结果显示,过表达SLIT1 3′UTR的HAECs中,miR-128-3p,-34a-5p,-27b-3p水平显著降低(P<0.05,P<0.01, Fig3B)。
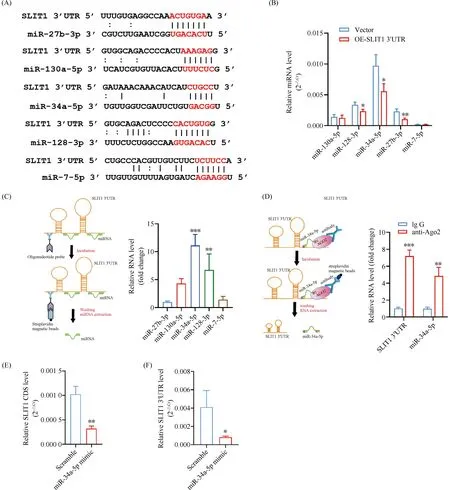
Fig.3 SLIT1 3′UTR served as a molecular sponge for miR-34a-5p
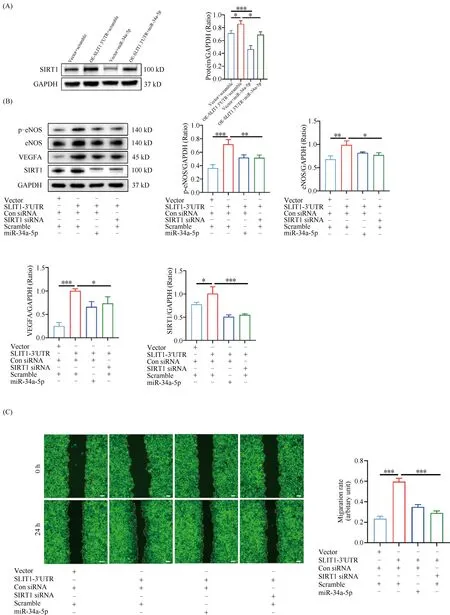
Fig.4 SLIT1 3′UTR promoted the migration and tube formation activities of endothelial cells by targeting miR-34a-5p/SIRT1 axis
先行在HAECs中过表达SLIT1 3′UTR,利用靶向SLIT1 3′UTR特异的生物素标记寡核苷酸探针富集HAECs中SLIT1 3′UTR,提取RNA并检测SLIT1 3′UTR结合的相关miRNA水平。RAP的结果显示,SLIT1 3′UTR可有效地结合miR-128-3p和miR-34a-5p (P<0.05P<0.01, Fig3C)。
为了进一步证实SLIT1 3′UTR与miR-34a-5p的结合作用,利用Ago2抗体进行RIP检测,结果显示,Ago2能够同时下拉到miR-34a-5p和SLIT1 3′UTR (P<0.01,P<0.001, Fig3D),进而明确了SLIT1 3′UTR和miR-34a-5p间的结合作用。RT-qPCR结果显示,在HAECs中转染miR-34a-5p能分别有效降低SLIT1 CDS和SLIT1 3′UTR水平(P<0.05,P<0.01, Fig3E,3F)。
2.4 狭缝引导配体1 3′UTR通过结合miR-34a-5p上调SIRT1调节内皮细胞表型
既往研究中,我们已经证实SIRT1是miR-34a-5p的下游靶基因[11],Western 印迹结果显示,转染miR-34a-5p 的HAECs中SIRT1表达显著降低(P<0.001, Fig 4A),并能有效抑制过表达SLIT1 3′UTR引起的HAECs中SIRT1升高(P<0.05, Fig 4A)。
为了进一步明确下游靶基因SIRT1对内皮细胞表型的调节作用,本文利用siRNA技术敲减HAECs中SIRT1表达,在HUVECS中过表达SLIT1 3′UTR,再分别转染miR-34a-5p和靶向SIRT1的siRNA,检测miR-34a-5p和SIRT1 siRNA(si-SIRT1)对过表达SLIT1 3′UTR的HUVECS表型的影响。Western 印迹结果显示,miR-34a-5p和si-SIRT1均一致性的抑制过表达SLIT1 3′UTR诱导的HAECs中p-eNOS、eNOS、VEGFA和SIRT1的水平升高(P<0.05,P<0.01,P<0.001 Fig. 4B)。划痕和血管内皮细胞成管腔结果显示,miR-34a-5p和si-SIRT1均可有效抑制SLIT1 3′UTR促进内皮细胞迁移(P<0.01, Fig. 4C)和血管内皮细胞成管腔作用(P<0.05,P<0.01,P<0.001, Fig. 4D)。
3 讨论
3′UTR是mRNA的非编码区[12],与酵母细胞相比,人类3′UTR的长度比酵母细胞高出10倍,而编码区的长度在两者之间却是相似的,这提示3′UTR在高等生物中发挥关键作用[13]。越来越多的研究表明,3′UTR介导了多种疾病发生过程,包括新冠肺炎,强直性肌营养不良和癌症等[14-16]。本文发现,人心肌中SLIT1 3′UTR水平显著高于SLIT1 5′UTR和SLIT1 CDS,该结果与以往发现心衰病人心肌中Ckip-1 3′UTR高于Ckip-1 CDS水平的报道[8]相似,但具体调节机制尚不清楚。
在生理性心肌肥厚中,微血管新生速度和心肌肥厚的程度相适应,在病理性心肌重构中虽然伴有微血管的新生,但是血管新生速度远低于心肌细胞对血供需求增加的速度[17,18]。血管生成在缺氧损伤后迅速发生,而内皮功能的调节对于维持新生血管稳态至关重要[19]。本文证实,HCM病人心肌中SLIT1 3′UTR表达降低,在功能上,SLIT1 3′UTR具有促进血管生成相关基因eNOS和VEGFA的表达,以及促进血管内皮细胞的迁移和成管腔能力,提示SLIT1 3′UTR具有促进血管再生的作用,通过提高SLIT1 3′UTR水平可能有利于改善血运重建,减轻心肌肥厚。
本文通过生物信息学分析预测到了5个可能与SLIT1 3′UTR有结合作用的miRNA,通过RAP和RIP实验证实了SLIT1 3′UTR与miR-34a-5p和miR-218-3p间特异性的结合作用。定量检测结果显示,HAECs中miR-34a-5p表达水平和SLIT1 3′UTR结合miR-34a-5p的能力均一致高过miR-218-3p,因此,本文重点关注SLIT1 3′UTR是否通过结合miR-34a-5p发挥对HAECs的表型调节作用。既往研究证实,抑制miR-34a-5p可上调ZEB1(Zinc finger E-box binding homeobox 1)表达,逆转缺氧诱导的JAK/STAT和PI3K/AKT通路中磷酸化激酶的减少,从而保护心肌细胞免受缺氧诱导的细胞损伤[20]。本文报道了SIRT1是miR-34a-5p的下游靶基因[11],本文结果证实,SLIT1 3′UTR可以作为分子海绵吸附miR-34a-5p,并特异上调SIRT1表达。
内皮SIRT1缺失或SIRT1失活经常伴随着许多心血管疾病[21]。Das等人发现,烟酰胺单核苷酸(NMN)可以增强SIRT1的活性,上调SIRT1的表达,促进血管生成并提高毛细血管的密度来改善老年小鼠的血流量,同时提高耐力[22]。eNOS主要在内皮细胞中表达,是细胞功能的中枢调节因子,参与维持内皮稳态,保持血管扩张,控制血压,并具有许多其他血管保护作用[23,24]。有研究证实,SIRT1可与eNOS直接相互作用,并激活eNOS[25,26],与之相符,本文发现,miR-34a-5p和SIRT1 siRNA可降低HAECs中eNOS和p-eNOS水平,而过表达SLIT1 3′UTR能显著升高eNOS和p-eNOS水平。因此,本文证实miR-34a-5p/SIRT1轴介导了SLIT1 3′UTR发挥促进血管内皮细胞迁移和管腔生成的作用。
综上所述,本文证实SLIT1 3′UTR可以特异性吸附miR-34a-5p,通过miR-34a-5p/SIRT1轴促进内皮细胞增殖、迁移与血管新生。在后续研究中,我们将进一步在整体动物水平研究SLIT1 3′UTR促进血管新生的作用机制,为进行基于3′UTR为干预靶点的心肌肥厚的治疗研究提供科学资料。
