核酸条形码技术:扩展蛋白质-蛋白质相互作用检测通量的新方法
2024-04-23李林鑫秦晓红米立志
李林鑫, 秦晓红, 米立志
(天津大学生命科学学院结构与分子生物学系, 天津 300072)
细胞通过蛋白质-蛋白质相互作用进行交流通讯和信号转导,并根据此调节细胞代谢、增殖、分化等众多生命活动[1]。针对细胞内蛋白质相互作用的研究正成为系统生物学研究的重要任务之一[2]。鉴于蛋白质-蛋白质相互作用在细胞通讯及细胞信号转导过程中所发挥的关键作用,绘制并阐明蛋白质-蛋白质相互作用网络图谱有助于揭示疾病发病机制,开发新型药物靶点[3]。
迄今为止,大量的蛋白质相互作用研究方法相继被开发,但不同方法各有利弊,数据质量参差不齐。低通量实验方法通常可获得高质量蛋白质-蛋白质相互作用数据,但碍于成本及实验流程等原因,难以用于大规模蛋白质-蛋白质相互作用网络研究。高通量的蛋白质-蛋白质相互作用检测方法[4]可同时检测逾数十万对蛋白质-蛋白质相互作用,但由于实验样本量的增加,这一类方法实验流程繁琐,技术门槛较高。
得益于下一代测序技术(next-generation sequencing, NGS)飞速发展,核酸条形码技术应运而生。核酸条形码技术[5]是将寡核苷酸标签附加在蛋白质、细胞、有机大分子和无机小分子等材料上,用在基因组结构测定[6]、蛋白质翻译活性的综合分析[7]、细胞命运谱系追踪[8]和药物靶点筛选[9]等领域。同时,核酸条形码技术也为高通量检测蛋白质相互作用提供了新的思路。目前,大规模的蛋白质-蛋白质相互作用检测方法主要可分为3类:并行一对一检测、一对多检测和多对多检测[10],这些方法大多依赖核酸条形码对目标蛋白质进行标记。
尽管在不同类型的蛋白质-蛋白质相互作用检测方法中,核酸条形码技术的应用方法不尽相同,但其设计原理具有高度的相似性。核酸条形码技术在蛋白质相互作用检测方法中的通用策略如Fig.1所示,本文凝练了核酸条形码的设计原则,归纳总结了其纠错策略、生成递送和读取流程。通过回顾核酸条形码技术在大规模的PPI检测方法中的经典应用,讨论了各种PPI检测方法的可靠性、成本及操作简便性。在此基础上,基于核酸条形码技术和NGS技术的发展现状,本文探讨了PPI检测方法的发展趋势。
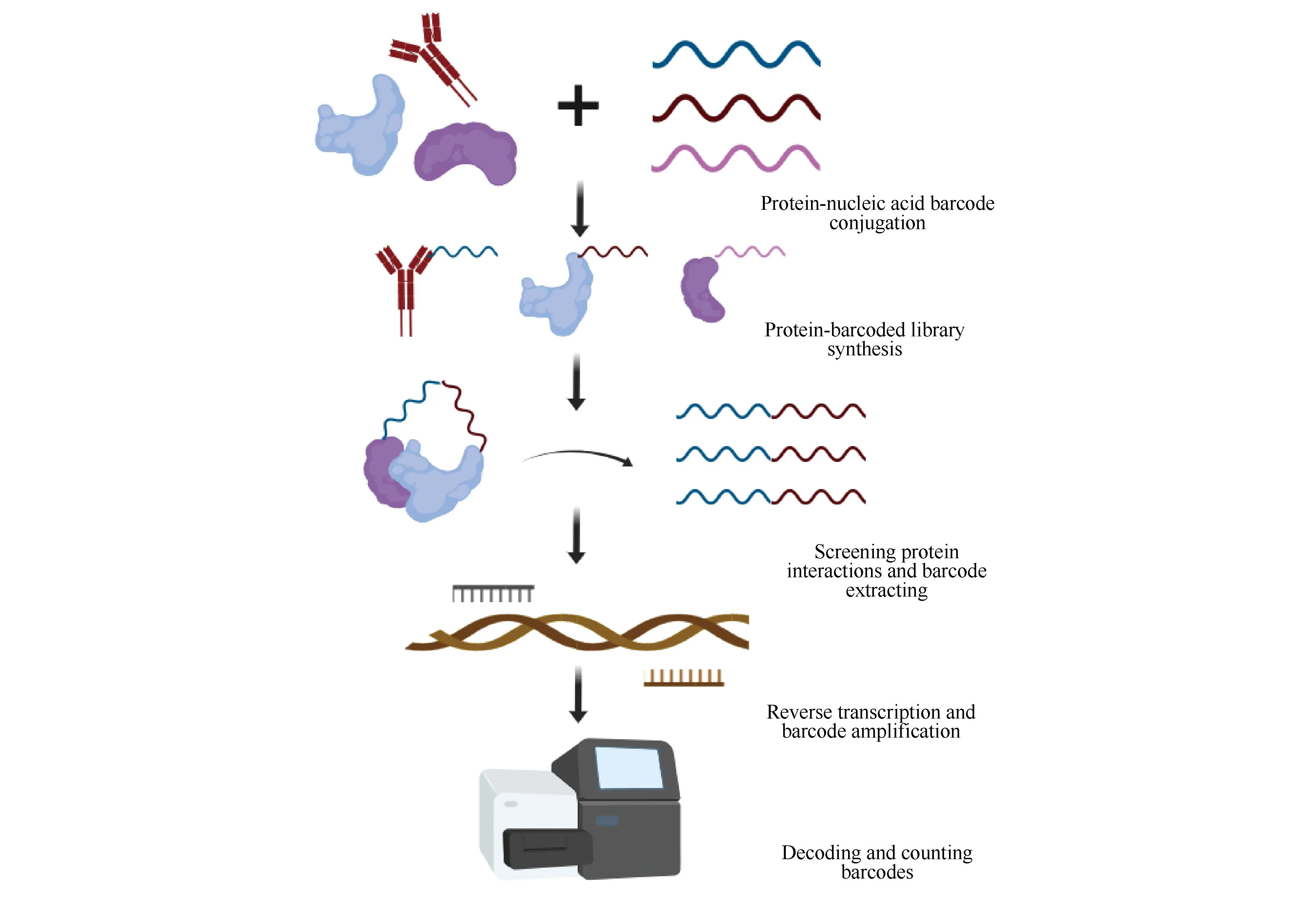
Fig.1 Common strategy for protein-protein interaction detection via nucleic acid-barcoding technologies
1 核酸条形码的设计
核酸条形码的设计是这一技术的运用基础,也决定着后续蛋白质相互作用检测数据的评价方式。核酸条形码的设计可以分为设计原则、生成递送和读取3个方面。其中,设计原则决定着条形码的多样性,即能够编码目的蛋白质的最大数量,而设计原则又会引申出条形码的合成与读取错误;条形码的生成和递送是指核酸条形码文库与目的蛋白质一一联系起来,其中不同的连接方式各有特点;而条形码的读取则是整个蛋白质相互作用检测的最后一步,阳性结果的条形码经过一定处理后读出结果,就能定性定量分析蛋白质相互作用。
1.1 核酸条形码的设计原则
在不同类型的蛋白质-蛋白质相互作用检测方法中,核酸条形码序列均是由随机或半随机的短核苷酸生成[11]。完全随机的核酸条形码理论上编码的数量为4n,其中n是核苷酸序列的长度。因此,随机的10 bp条形码理论上会编码的多样性达410(~106)。以Yachie等人[12]开发的条形码融合遗传酵母双杂交(barcode fusion genetics yeast 2 hybrid,BFG-Y2H)技术为例,在每个“猎物”蛋白质和“诱饵”蛋白质基因编码区前,设计了两个25 bp的随机核酸条形码区域。酵母杂交后,在Cre重组酶的作用下,相互作用的“猎物”蛋白质和“诱饵”蛋白质的基因编码区前,紧邻的1个核酸条形码被交换。因此,通过测序鉴定相互作用的蛋白质对,这一方法理论上可以研究2×449个不同的蛋白质-蛋白质相互作用。随机核酸条形码编码多样性丰富,理论极限值仅受条形码长度限制,设计简单,合成成本低;但可能产生的连续poly-N区域会对寡核苷酸合成、PCR扩增和测序造成影响[13],而非恒定的GC比例也影响着不同条形码的PCR扩增。此外,部分位于读码框内的随机条形码会影响目的蛋白质表达。
为了避免上述情况,核酸条形码采用半随机设计[14],将条形码中一些特定位置的核苷酸限制为一个或多个指定的核苷酸。这样的设计以牺牲条形码编码多样性为代价来维持恒定的GC比[15],或是通过增加编码长度来避免连续的poly-N出现。
无论是采用随机或是半随机的方法,在设计过程中都会面临两大局限问题。其一,由于递送效率和设计的要求,均需要保证条形码具有数量上的冗余,而这就会导致测序(条形码读取)信息的重复[16],显著影响系统的检测效率。其二,当前大多数蛋白质相互作用检测系统在条形码的读取中都是利用通用引物对编码序列进行扩增和测序,不能查询特定相互作用蛋白对的信息。
解决这些问题的最好方案是赋予每组条形码一个特定的“访问地址”。在设计条形码时将待编码蛋白质分为若干组,并将条形码的编码区分为可变区和恒定区两部分:可变区是随机的,发挥分子标识符的作用;而恒定区是固定的,用于编码特定组别的蛋白质,以便在检测相互作用时随机访问该组特定条形码。Lee[17]等人正是利用这一思路开发了冗余优化的编码方案,他们设计的条形码长度约为150 bp,左右两端各有20 bp的“访问地址”序列作为PCR引物结合位点。为了实现稳健的随机访问,他们还构建了有效的PCR引物来扩增特定的条形码序列,这些引物具有以下几个特性:(1)不易形成二级结构和引物二聚体;(2)不含长段的均聚物;(3)熔解温度限制在一个狭窄的范围(55~60 ℃)。与其他引物相比,这些引物序列具有至少30%的独特性,保证了访问的特异性。
除此之外,核酸条形码的合成和读取也易出错误,其中插入、缺失和替换等错误的频率在1%左右[18]。因此,在当前合成与检测技术条件下,如何纠错显得至关重要。在核酸条形码发展的初期,合成的条形码通常需要被克隆到大肠杆菌或酵母的质粒中,通过 Sanger 测序进行验证。这些步骤昂贵耗时,难以实现自动化。对于检测插入或缺失突变,会将合成的序列与编码抗生素耐药性的筛选标记或荧光蛋白质连接在一起[19]。当条形码存在插入或缺失突变时会导致编码序列移码,使报告基因失活。当前核酸条形码的验证通常采用高通量测序的方法[20],相比便携式的测序仪如 MinION ,采用 Illumina HiSeq 和Mi Seq等高端测序仪可以减少测序误差。因保守估计,MinION[21]测试错误率可高达30%。Matzas等人[22]则开发了 Mega cloning 技术,利用 GS FLX测序平台结合自动化的移液管,实现对寡核苷酸文库的测序和自动分类,便于随机访问感兴趣的条形码并保证了极低的测序错误率,但其成本与技术门槛较高,无法短时间内大规模推广。Yazdi等人[23]则通过改进编码、储存、读取、纠错等数据处理流程,降低了测序的成本,并在使用 MinION 的基础上,使测序错误率降低至0.02%。在编码步骤中,他们设计的条形码同样包含了“访问地址”和“编码区”,便于随机访问,其长度可达1 000 bp,并通过专门的受限编码技术平衡了每个条形码的GC含量;在读取纠错阶段,他们采用了共识对齐算法,将测序阶段引入的插入和替换错误“转换”为删除错误,然后通过对符合长度的高质量读取进行多序列比对,找出共有序列,纠正测序错误。
1.2 核酸条形码的生成和递送
核酸条形码文库的合成非常高效,以现有的合成技术在短时间内生成包含数十亿个条形码的文库[24]。但递送条形码的方式则各有特点,目前,主要可分为分子克隆、体外展示和蛋白质标签法等3类。
1.2.1 分子克隆方法 分子克隆方法主要指将合成的条形码文库通过Gibson组装[12]、Gateway克隆[25]、酶切连接或者重叠PCR[26]等方法构建到表达目的蛋白质的质粒上(见Fig.2A),在基因水平上将二者连接起来。这些构建方法对条形码文库通常有一定要求,例如条形码长度尽量短、 GC 比例合适、条形码上下游拥有通用引物接头等。此外,在部分酵母双杂交方法中直接将蛋白质的编码序列用作分子条形码,在PPI筛选结束后通过测定目的蛋白质编码序列,来识别相互作用的蛋白质对[27, 28]。
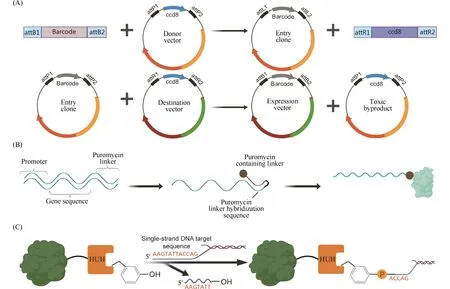
Fig.2 Several delivery methods of nucleic acid barcodes
然而,上述分子克隆方法通常需要借助Cre、PhiC31等基因重组酶将条形码连接到表达目的蛋白质的质粒上,步骤繁琐且容易出错,同时生成的条形码序列相对较长,读取成本高。例如,Yu等[29]开发的 Stitch-seq 方法是利用两轮 PCR 将相互作用的蛋白对的2个开放读码框“缝合”在一起生成条形码,然后通过NGS测序读取条形码,这一设计虽然精简了系统设计,但串联扩增子的长度显著大于当前NGS测序仪可读取的有效长度。Yang等人[30]开发的rec-YnH (recombination yeast n hybrid, rec-YnH)方法,则是利用同源重组原理将表达“猎物”蛋白质和“诱饵”蛋白质的质粒融合,融合后的质粒经提取并通过Covaris酶裂解成为线性化片段,随后通过分子内连接环化,经两轮PCR扩增后获得正反相连的2个编码相互作用蛋白质开放读码框的DNA片段,将这一DNA片段作为识别PPI的条形码。
1.2.2 体外展示方法 在核糖体展示(ribosome display)[31]、mRNA展示(mRNA Display)[32]等技术中,通过一些简单的生化反应就能将核酸条形码与目的蛋白质连接起来,在体外生成蛋白质编码文库(见Fig.2B)。在核糖体展示技术中,目的蛋白质编码基因的终止密码子缺失,或通过添加核糖体停滞剂以稳定肽基-tRNA连接,核糖体无法从新生肽链脱落,最终使目的蛋白质与对应的mRNA分子一起与核糖体形成稳定复合物。这项技术简单高效,单次实验可以生成约1014个蛋白质-条形码复合物,产生的文库大小仅受溶液中核糖体数量的限制[33]。在后续分析中,蛋白质的编码mRNA被直接用作核酸条形码。基于核糖体展示,Gu等人[26]开发了单分子相互作用测序技术(single-molecular interaction sequencing,SMI-seq),通过在体外转录时生成mRNA-cDNA杂交文库,之后以杂交文库中的mRNA分子为模板,体外翻译形成蛋白质-核糖体-mRNA-cDNA的复合物,用于蛋白质相互作用分析。这类方法优点突出,缺点同样明显。首先,体外检测方法无法测定依赖翻译后修饰或定位的蛋白质相互作用;其次,分子量较大的蛋白质目前无法应用于体外展示系统;最后,mRNA分子易降解,如何形成稳定的蛋白质-核糖体-mRNA复合物依然是这一类技术的关键。
1.2.3 蛋白质标签法 除上述两种方法外,还可以利用SNAP Tag(约19 kD)[34],Halo Tag(约33 kD)[35]、和CLIP-tag(约20 kD)[36]等基因工程酶将核酸条形码和目标蛋白质共价连接,以用于在抗体筛选、肿瘤标志物检测等领域。Gordon等人[37]最近报告了另一种DNA-蛋白质偶联策略,原理正如Fig.2C所示,他们利用HUH核酸内切酶结构域(约10~30 kD)切割特定的单链DNA序列,产生一个游离的3′-OH基团和一个与ssDNA共价连接的中间体,利用这一方法,就可以将目的蛋白质(与HUH核酸内切酶结构域相连的)与单链DNA共价连接起来。这种方法的一个主要优点是它不需要制备与DNA连接的配体,就可以直接用未修饰的DNA链生成条形码-蛋白质文库。同时这类方法也具有自身的局限性,首先,核酸条形码的部分序列是固定的,以此满足酶切的要求,这会导致核酸条形码多样性受限;其次,目的蛋白质需要与酶共表达后纯化或单独纯化后再与特定酶连接,这一过程较其他标记方法增加了额外的工作量;第三,部分酶的工作条件苛刻,势必会导致与之相连的蛋白质在核酸条形码标记过程中变性,影响后续的相互作用的检测。
然而,不同的基因工程酶的标记效率也有所不同,有研究表明,SNAP Tag[38]的标记效率约为61%~86%,而Halo Tag的标记效率为55%~66%。不均一的标记会导致文库游离大量寡核苷酸,对后续实验产生影响,因此,标记后产生的文库需要纯化。一些研究[39]中直接使用限制性内切酶消化游离的寡核苷酸,从而纯化文库,大多数方法采用亲和层析[26]的方法纯化出蛋白质-核酸条形码复合物文库后,再使用限制性内切酶消化,这样的方法能够有效去除游离的寡核苷酸,但无法保证文库质量,即文库中会存在部分未被标记的蛋白质。这虽然会导致部分假阳性结果,但由于这部分蛋白质未标记核酸条形码,在定量分析相互作用时反而不会被统计,从而可以在二次筛选中去除假阳性。
1.2.4 小结 上述的各种方法均有其各自的优势和不足,在实际操作中,应根据目的蛋白质的大小、性质等因素选取最适合的方法。值得注意的是,选择上述两类递送方法时,还应考虑核酸条形码的长度对相互作用蛋白质检测的影响。在分子克隆方法中,核酸条形码仅在基因水平上“代表”目的蛋白质或特定的相互作用反应,而不与蛋白质本身产生任何形式的连接。因此,在这类方法中,核酸条形码的长度不会对相互作用蛋白质产生任何影响。然而,在生化反应方法中,类似蛋白质标签,核酸条形码直接连接在目的蛋白质上,其长度直接影响着目的蛋白质的构象,甚至在目的蛋白质相互作用时产生空间位阻,进而影响相互作用的检测。同时,连接在1个待测蛋白质上的核酸条形码还有可能与其他目的蛋白质相互作用,导致假阳性的结果。以Kara等人[10]开发的蛋白质-蛋白质相互作用测序(protein-protein interaction sequencing,PROPER-seq)技术为例,首次实验中就有13组PPI数据同样在阴性对照实验中被检测到,基本可以确定是较长的mRNA条形码与蛋白质相互作用所导致的假阳性。
1.3 核酸条形码的读取方法
条形码的读取经历了从早期桑格测序[40]、微阵列检测[26]到如今的高通量测序[30]的逐渐进步历程,这些方法均包括以下流程:(1)条形码预处理;(2)核酸提取、条形码扩增;(3)测序鉴定。测序流程如Fig.3 所示,Galinski等[25]基于分裂TEV酶方法开发的GPCR活性分析技术中,每一个独特的相互作用会表达不同的标签(expressed oligonucleotide tag,EXT),而这些表达标签由独特的条形码序列编码,从而形成报告RNA,在诱导发生蛋白质相互作用后,通过提取细胞内总RNA,逆转录编码表达标签的报告RNA,经高通量测序定量定性分析报告RNA的表达,以鉴定蛋白质的相互作用及其强度。这一方法定量检测蛋白质-蛋白质相互作用强度,但受限于表达标签的设计,实验通量有限,若想提高通量则需要赋予每个PPI一个独特的编码标签,工作量较大。

Fig.3 Schematic representation of GPCR split TEV assays
同时,每种读取方法都会受到条形码检测错误的影响,除设计原则中提到的合成和高通量测序中出现的错误外,读取流程中还可能由于PCR扩增及条形码预处理引入错误[41],包括碱基的缺失、插入和替换等错误[42]。例如,蛋白质相互作用测序技术(protein-protein interaction sequencing,PPiSeq)[43]的读取结果,就会严重受到条形码预处理的影响。此技术的检测原理是在编码2个目的蛋白质的条形码前后分别放置了loxP位点,蛋白质相互作用会引发重组,从而介导2个编码目的蛋白质的质粒进行分子间重组,2个条形码由此被连接在一起。然而,这一过程中可能导致大量编码非相互作用蛋白质的质粒发生重组,在PCR时被扩增,影响定量分析。这一重组过程是可逆的,为读取检测带来了压力。总之,应当尽量精简读取之前对条形码进行操作,预处理工序越复杂,出现的错误可能就越大。Ullal等[44]开发的光裂解DNA条形码标记抗体技术(antibody barcoding with photocleavable DNA,ABCD)提供了一种很好的解决办法,抗体与DNA条形码之间通过一种光降解接头相连。将抗体-条形码文库与目的细胞共同孵育后收获细胞,光照切割连接子释放唯一的DNA条形码,之后利用荧光杂交技术定性定量分析PPI,省去了PCR扩增等步骤,显著增加了数据可信度。然而,其条形码多样性受到一定限制,因为,这些条形码需保证不会与人类基因组发生交叉反应。
2 核酸条形码的应用
自核糖体展示以来,核酸条形码在蛋白质-蛋白质相互作用检测中的应用已近30年。随着合成和测序技术的进步,条形码设计越来越简洁、多样性、递送方式与读取方式也变的更多样、方便和低廉。Table 1展示了几类具有代表性的核酸条形码在PPI检测中的应用及其特点。这之中,本文根据不同的PPI检测方法特点将其分为3类:并行一对一检测方法、一对多检测方法和多对多检测方法。这些方法各具优势又有其自身的不足之处,下文将对这几类应用场景进行逐一讨论。
2.1 核酸条形码在并行一对一蛋白质相互作用检测方法中的应用
并行一对一检测方法来源于酵母双杂交(yeast 2 hybrid, Y2H)[45]、蛋白质片段互补分析(protein complementary assay, PCA)[46]等一对一检测方法,这类方法单次只能检测一对蛋白质的相互作用,通过特定报告基因的表达,以检测相互作用的强弱。而并行一对一检测方法则是以此为基础,通过并行的方式同时检测多对蛋白质的相互作用。这一方法虽然操作简单和结果直观,但检测通量严重不足,且无法检测三元或多元复合蛋白质之间的相互作用。
将核酸条形码技术与并行一对一检测方法结合能大幅提升检测通量。Marc Vidal实验室开发的Stitch-seq技术[29],将2个蛋白质的开放阅读框作为核酸条形码来确定相互作用的蛋白质对,实现了大规模并行一对一检测。这一方法虽然提高了相互作用蛋白质筛选的规模和灵敏度,但未能充分发挥核酸条形码技术在定量分析和检测方面的潜力[47]。
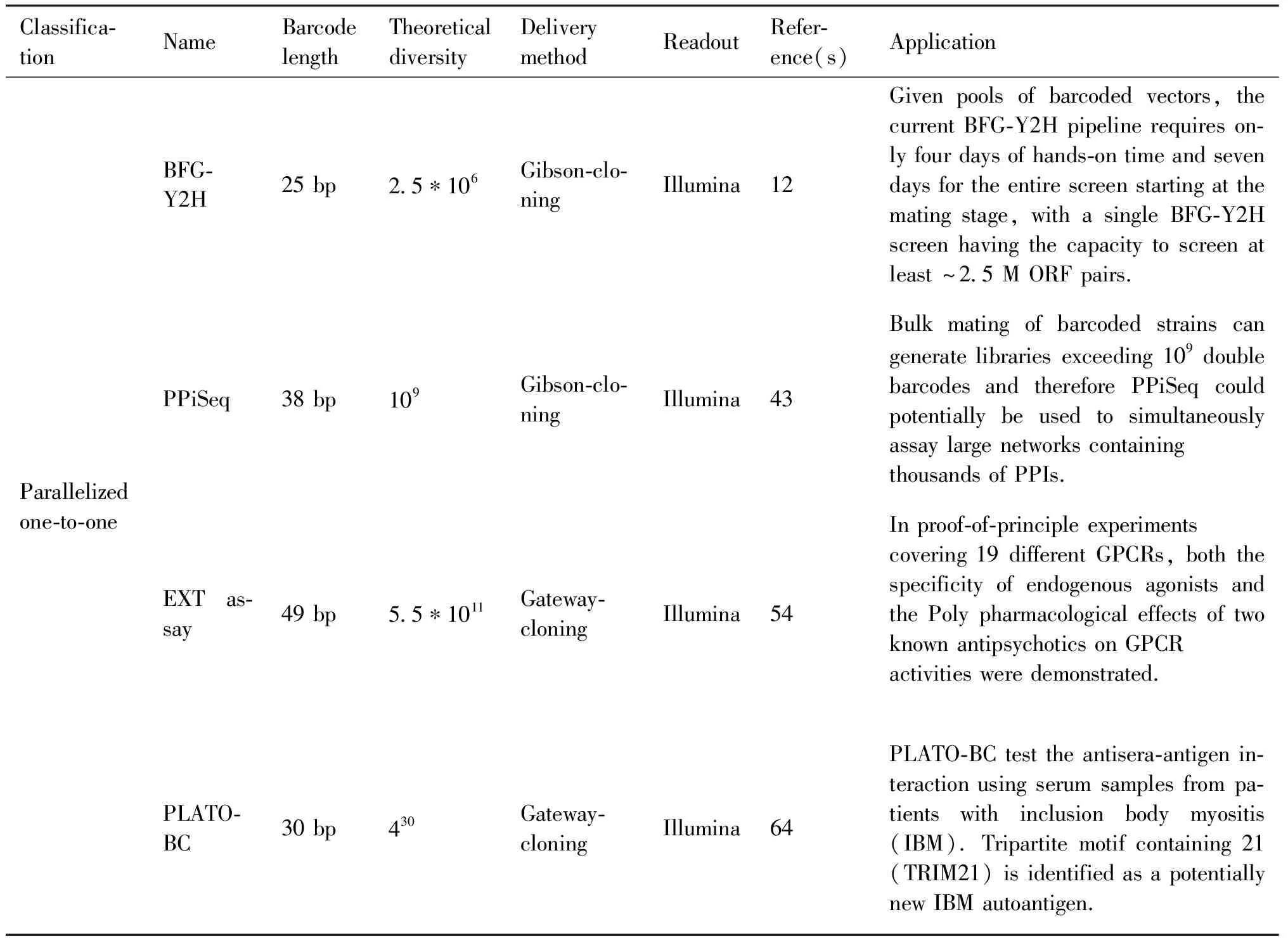
Table 1 Overview of barcoding techniques in PPI detections and their properties
BFG-Y2H技术[12]也是核酸条形码技术在蛋白质相互作用检测中较早的应用。BFG-Y2H基于酵母双杂交技术构建“猎物”和“诱饵”质粒文库,每个质粒上表达与其蛋白质对应的两个特异条形码。“猎物”质粒上第一个条形码(X-BC1)和“诱饵”质粒第两个上条形码(Y-BC2)的两侧,存在有定向特异性的loxP和lox2272重组位点。当两个酵母文库杂交后,Cre重组酶诱导分子间交叉反应,使两个质粒之间物理条形码交换。并利用选择性培养基上筛选存在相互作用蛋白质对的酵母细胞,之后通过测序确定相互作用的蛋白质对。相较于Stitch-seq等技术,BFG-Y2H创新性的利用较短的核酸条形码来标记蛋白质,降低了测序成本。同时该方法不仅定性分析蛋白质相互作用,还能定量表征其相互作用强度,显著提升了筛选的效率。同时,由于其条形码质粒文库具有通用性,原则上其Cre介导的条形码融合技术能应用于其他蛋白质相互作用检测。例如BFG-PCA[48]、BC-PCA[49]和BFG-GI[50]等技术。此外,BFG-Y2H技术还用于其他特定的研究。Schor等[51]利用BFG-Y2H技术筛选出了BIKE磷酸化的下游靶点网格蛋白相互作用体1(clathrin interactor 1,CLINT1),揭示了CLINT1被磷酸化后介导登革热病毒感染的机制。Celaj等[52]优化了BC-PCA技术,并将其用于酿酒酵母中,测量了14种环境下1 379种蛋白质二元复合物的体内丰度,揭示了酿酒酵母的某些自适应调节策略。
除与酵母双杂交技术连用外,核酸条形码技术也广泛用于其他并行一对一检测方法的改良和优化。Ulrich等[43]基于PCA技术,开发了类似BFG-Y2H的PPiSeq技术。他们将“猎物”和“诱饵”蛋白质与分裂的小鼠二氢叶酸还原酶(mouse dihydrofolate reductase, mDHFR)融合,并在质粒上构建特异性条形码。条件性诱导表达的Cre重组酶构建在“猎物”质粒上。当2个蛋白质相互作用时,mDHFR被重组,使菌落能够生长在含有酿酒酵母DHFR抑制剂甲氨蝶呤的培养基上,Cre重组酶介导的基因重组将2个条形码整合到同一染色体上。相比BFG-Y2H技术,依赖于PCA的PPiSeq技术在内源性启动子的作用下,表达整合在基因组上的融合蛋白质,蛋白质产物在细胞内的自然生理环境中相互作用,是检测动态PPI的可行性平台[53]。Anna等人[54]将分裂的烟草蚀刻病毒蛋白酶与核酸条形码相结合,开发了表达寡核苷酸标签的分析系统(expressed oligonucleotide tag assay,EXT assay)。在这一体系中分裂的TEV蛋白酶分别与“猎物”或“诱饵”蛋白质相连,末端连接转录激活因子(gal4-vp16, GV)。当2个蛋白质存在相互作用时,分裂的TEV酶重组为有活性的蛋白酶,切割释放末端的GV入核以激活寡核苷酸标签的转录,通过测定逆转录EXT的产量与序列就能确定相互作用的蛋白质对及其强度。除此之外,核酸条形码技术还能分裂蛋白生物传感器(split biosensor assay,SBA)技术[55]结合使用,这也是大规模并行一对一检测方法开发新的发展趋势。因为SBA是利用蛋白质-蛋白质相互作用诱导2个先前无功能和无结构的蛋白质片段重新折叠形成一个有功能的蛋白质作为报告因子,因此,相较于利用共定位检测的传统的酵母双杂交技术,SBA能稳健的检测动态的PPI,且信噪比通常更好[56]。
核酸条形码技术与酵母双杂交或分裂蛋白质生物传感器等技术连用,仅仅提升了其实验通量或定量分析能力,但无法改变并行一对一检测方法的固有缺陷,例如无法同时检测多蛋白质复合物的相互作用。
2.2 核酸条形码在一对多蛋白质相互作用检测方法中的应用
一对多检测方法一般是通过亲和纯化[57]、蛋白质微阵列[58]等方法来识别共纯化的蛋白质是否具有相互作用。这一类方法的另一个基础是体外翻译技术(invitrotranslation,IVT)。Nirenberg和Matthaei[59]于1961年首次报道了体外翻译过程,该过程利用细胞裂解物或纯化的蛋白质翻译复合物,实现了蛋白质的体外合成。IVT技术一经推出,就立刻被应用于蛋白质相互作用的检测。区别于当时盛行的酵母双杂交技术,基于IVT开发的核糖体展示等技术在体外合成蛋白质的过程中,蛋白质产物与其对应的mRNA结合在一起。这一技术有助于在体外大规模、一锅合成蛋白质-核酸条形码文库,成为表征生物信号通路和发现候选生物药物的重要工具[60]。
核糖体展示检测蛋白质相互作用的工作流程[61]为:首先是将文库与感兴趣的固定化多肽或蛋白质进行孵育,洗去无相互作用的成员,纯化结合的成员,将mRNA逆转录为cDNA。然后,通过PCR扩增、测序来确定结合的蛋白质。最初,核糖体展示依赖于桑格测序,单次实验可检测的序列数仅有102~103条左右。随着NGS技术的发展,分析的序列数量增加10 000倍以上(超过107条),显著提高了实验的通量并降低了成本[62]。Elledge等[63]开发的翻译开放阅读框并行分析技术(parallel analysis of translated open reading frames,PLATO)是在核糖体展示的基础上,将体外合成的蛋白质-mRNA文库与固定化的“诱饵”蛋白质共同孵育,最后逆转录富集的文库成员的 mRNA 并进行测序,确定相互作用的蛋白质。通过将表皮生长因子受体 (epithelial growth factor receptor, EGFR) 家族的17个预测靶标蛋白质使用PLATO展示后,再与偶联Gefitinib的磁珠共孵育,Elledge等人成功检测到了10个显著富集的靶标蛋白质。这一结果证明,PLATO在花费和时间成本上具有非常明显的优势,并且跟所有体外展示技术一样,能够非常好的应用于抗体开发和小分子化合物的构效关系分析。然而,利用蛋白质开放阅读框作为条形码不仅需要在测序前进行多次预处理,而且某些较长的条形码无法一次测通,影响了系统的精确性。因此,Kong[64]等优化了 PLATO 技术,在每个蛋白质ORF的3′端添加了条形码与蛋白质一一对应,简化了测序前的处理工作,提升了系统的效率。值得注意的是,核糖体展示技术的一个关键点是核糖体稳定缓冲液需要与核糖体融合蛋白质兼容。当这一因素影响了核糖体展示的使用时,mRNA展示可能是一个合适的替代方法。
类似核糖体展示,mRNA展示也能用于在IVT期间将蛋白质与其mRNA连接起来,但与核糖体展示不同,mRNA展示是利用嘌呤霉素融合到mRNA的3′端,产生共价连接的多肽-核酸复合物,并从核糖体中分离出来用于下游文库的构建。1997年,Szostak等[65]首次报道了这一技术。他们通过DNA连接子将嘌呤霉素融合到mRNA的3′端。经核糖体翻译将杂交mRNA片段翻译成相应的多肽,当延伸到3′ DNA片段时,核糖体停止翻译,3′嘌呤霉素进入核糖体的A位点,并与生长的多肽形成酰胺键,从而使mRNA-DNA-肽复合物从核糖体释放。相较于核糖体展示,嘌呤霉素连接的核酸多肽偶联物的稳定性较高,是研究短肽结构的理想选择。随后,Philip等[66]将mRNA展示应用于蛋白质相互作用筛选研究中,筛选出了几个与抗凋亡蛋白质Bcl-XL相互作用的蛋白质,证实了这一技术的巨大潜力。mRNA展示对300个氨基酸长度以下的多肽最为有效[65],而核糖体展则是分析大蛋白质的首选方法。
随着NGS技术的长足发展,越来越多的一对多PPI检测系统开始倾向于使用短核苷酸条形码标记蛋白质,使其便于后续测序。同时,一些DNA-蛋白质偶联物的合成技术被相继开发,并用于目标蛋白质的外源性标记。Yazaki等[67]开发的Halo Tag条形码检测技术,使用高亲和力捕获标签Halo Tag将蛋白质与DNA寡核苷酸连接起来,随后通过高效液相色谱纯化DNA-Halo Tag-蛋白复合物。相对于其他标记方法,Halo Tag标记的蛋白质以1∶1比例与一个小的化学配体氯烷烃结合,因此,易于确定融合蛋白质分子的数量。Barber[68]等基于CRISPR技术开发的Cas9介导的自组装系统(peptide immobilization by cas9-mediated self-organization,PICASSO),利用sgRNA作为条形码标记蛋白质,之后纯化出sgRNA-dCas9-目的蛋白质复合物。将复合物文库与DNA微阵列共孵育,使dCas9能够在sgRNA引导下结合互补的DNA,从而将互作蛋白质固定,检测蛋白质-蛋白质相互作用。与噬菌体和核糖体展示相比,PICASSO技术规范了蛋白质水平的实验流程,对蛋白质相互作用进行可视化读取。并且,由于在设计时就确定了sgRNA与蛋白质的一一对应关系,根据sgRNA设计的DNA微阵列序列是固定的。因此,当把dCas9-sgRNA文库与dsDNA微阵列孵育几个小时后就可以进行PICASSO筛选,无需测序,避免了过高的测序成本和核酸读取相关的错误。上文提到过的ABCD 平台[44],创新性地将条形码和抗体用光照分解的连接子连接起来构建文库,之后将文库与特定细胞孵育来检测特定肿瘤标志物。ABCD平台灵敏度很高,能够在单细胞水平上研究临床样本,即使是p53结合蛋白1 (p53-binding protein 1,53BP1)和磷酸化组蛋白H2A突变体X(Phospho-H2A histone family member X, pH2A.X) 这样的稀缺蛋白质,也可以在单细胞水平检测到。利用这一技术对分离的罕见的细胞和克隆群体进行大规模的蛋白质定位,以深入研究癌症的异质性、耐药性和循环肿瘤细胞的临床应用[69]。
一对多检测方法在抗体定向进化、药物靶点筛选等领域被广泛采纳。但大多数一对多检测方法对蛋白大小有严格限制,无法在体外翻译纯化较大的蛋白质。此外,体外环境使得部分蛋白质无法实现正常的翻译后修饰,因此,无法正常发挥其功能,而且在非生理状态下某些PPI无法被正常检测,这些问题都有待后续关注和改进。
2.3 核酸条形码在多对多蛋白质相互作用检测方法中的应用
相对于并行一对一和一对多检测方法,多对多检测方法强调非二元性绘制PPI图谱[10],即单个实验不仅能检测到多对蛋白质相互作用,也能检测到多重蛋白质相互作用。单个被检测的相互作用有可能是一对蛋白质参与的,也有可能是3个或4个蛋白质相互作用形成的复合物。多对多检测方法的出现可以说完全依赖于NGS技术的发展,其实验规模的要求使得几乎所有多对多检测方法都采用核酸条形码来标记目标蛋白质。
目前,一部分多对多检测方法是在一对多检测方法的技术基础上开发的,通过改进条形码递送方式、优化PPI检测环境等手段,实现多对多检测的目的。例如上文所述SMI-seq[26]技术就是通过核糖体展示和Halo Tag技术,将含不同通用引物序列的DNA条形码分别连接到一组“猎物”和一组“诱饵”蛋白质上。两个文库的蛋白质发生相互作用后被固定到超薄的交联聚丙烯酰胺凝胶层中,然后利用桥式PCR对DNA条形码进行原位扩增,经DNA测序确定各位点的蛋白质相互作用信息。因为两组蛋白质所携带的DNA条形码的通用引物序列不同,所以利用不同的通用引物经两轮PCR扩增就能区分“猎物”蛋白质和“诱饵”蛋白质,实现了多对多蛋白质相互作用的检测。SMI-seq技术在单分子水平上检测蛋白质相互作用,每次反应可以同时检测高达上百万的PPI,既能定性又能定量分析蛋白质相互作用及其强度。但其技术门槛较高,操作繁琐,难以大范围推广。基于体外翻译展示系统,Johnson等[10]开发的蛋白质-蛋白质相互作用测序(protein-protein interaction sequencing,PROPER-seq)技术不仅简化了操作,同时相较SMI-seq检测通量也显著增加。PROPER-seq技术将单次PPI检测实验分为3个独立的模块:SMART-display、INLISE和Identification。SMART-display类似mRNA展示,将输入的一组细胞经历RNA提取、cDNA合成、模板转换反应、混合基因纯化、体外转录和翻译等通用步骤就可以得到包含整个细胞基因组的蛋白质-mRNA文库,输出的文库内容完全取决于输入的细胞。INLISE是PROPER-seq关键的第3步。首先,将SMART-display输出的文库中的一部分通过嘌呤霉素连接子序列上的生物素固定在素磁珠上,作为“诱饵”蛋白质文库,另一部分游离文库作为“猎物”蛋白质文库。两个文库的蛋白质发生相互作用后,将其mRNA转换成双链DNA,通过T4连接酶将相互作用蛋白质的cDNA连接,,形成cDNA1-linker-cDNA2形式的嵌合序列。最后,嵌合序列被添加测序通用引物。第3步扩增文库,进行NGS测序鉴定相互作用。PROPER-seq单次实验的通量十分惊人,且通过模块化各个步骤,实验人员仅需更换上游输入的细胞即可获得不同细胞的PPI网络,可推广性相当高。尽管如此,PROPER-seq仍有局限,例如蛋白质-DNA相互作用可能会引起的假阳性结果,各模块的操作繁琐、且技术门槛较高。相对而言,Payam等[70]开发的Abseq技术的操作更加简单,技术门槛也更低。他们利用约100 bp的 DNA条形码标识表达展示特定抗原的单细胞,同时用59 bp的DNA条形码标记抗体,将抗体文库与抗原细胞表面展示文库共孵育,通过液滴微流控技术进行单细胞分离,之后,通过重叠PCR将抗体条形码与细胞条形码拼接,测序确定抗体与抗原的识别。相较于传统抗体筛选方法,这一技术允许抗体识别经过修饰的生理状态下的抗原。其次,多对多的筛选特性使得Abseq在研究未知药物靶点和致病机制等方面具有较大潜力。目前,Abseq的通量和灵敏度主要受限于测序深度,同时条形码可能会在反应过程中丢失,重叠PCR也会引入新的错误。除此之外,文库制备成本也比较昂贵,为了使液滴PCR高效,液滴的体积和PCR试剂的浓度都是至关重要的[71]。
相互作用依赖PCR (interaction dependent PCR,IDPCR)在检测成本及技术上具有优势。IDPCR[72]技术是利用分子内和分子间形成双链DNA的熔解温度(Tm)的差异,实现在37 ℃下利用DNA聚合酶PCR扩增6-nt互补的分子内双链而非分子间形成的双链。设计上,可以将“猎物”和“诱饵”蛋白质分别连接到2个单链DNA条形码的3′端,当二者具有相互作用时,可使条形码上6-nt的互补区域可以形成分子内双链,因此,能够利用与“猎物”或“诱饵”蛋白质条形码对应的通用引物经PCR扩增出条形码。IDPCR操作简单,敏感度高,仅在n h内完成,相比其他类型多对多检测方法成本优势明显。之后,Mcgregor等[39]在此基础上开发了无需蛋白质纯化的相互作用测定(interaction determination using unpurified proteins,IDUP)技术。与IDPCR技术不同,IDUP技术是利用抗体识别靶向蛋白质,以检测靶标蛋白质与小分子配体的相互作用。未经纯化的蛋白质与携带DNA条形码的小分子配体和携带DNA条形码的抗体相互作用,形成蛋白质-抗体-配体三元复合物,其中抗体与配体上的2个DNA条形码可以通过PCR扩增测定。将IDUP技术应用于表达His6-Bcl-xL的HEK293 T细胞的裂解物中,成功检测出了Bcl-xL与内源性Bad之间的相互作用。这证明IDUP技术能够利用细胞裂解液来研究蛋白质-配体间的相互作用,因此,靶标蛋白质保留了天然的翻译后修饰,且与粗细胞裂解物兼容,实现对难以纯化、可溶性差、结构不稳定和易聚集的蛋白质进行相互作用检测。然而,细胞裂解液中,一些杂蛋白质可能会促进目的蛋白质与小分子配体的结合,从而导致假阳性增多。此外,IDUP技术还能应用于小分子化合物的构效分析。Alix[73]等将IDUP应用于DNA编码的小分子化合物和DNA标记的人类激酶文库,从中鉴定出了大量已知的小分子与蛋白质的相互作用,并揭示了乙基丙烯酸是MAP2K6激酶的新配体和抑制剂,能够通过非保守半胱氨酸残基的烷基化来抑制MAP2K6。
目前,多对多检测蛋白质-蛋白质相互作用的方法还处在发展的初期,各种方法不断涌现,包括重组酵母交配SynAg[74]、临近连接与双标签微阵列读出[75]等方法。但各种方法的可操作性、系统稳定性与实际表现仍有待进一步验证,但相信核酸条形码蛋白质标记技术仍将在未来很长一段时间是开发相互作用检测的重要工具。
3 问题与展望
核酸条形码技术促进了高通量、大规模蛋白质相互作用检测方法的发展,通过寡核苷酸作为待测蛋白质的分子标识,提高了PPI检测的通量。随着生物偶联化学和NGS技术的进一步发展,我们或许可以期待核酸条形码技术在蛋白质相互作用检测领域得到更普遍的应用,而这一期许有赖于以下方向的改进与突破。
首先,是条形码递送方式的改进。最近报道的阳离子脂质体介导的蛋白质递送方法正如Fig.4 A所示。该方法是利用mRNA展示将体外合成的蛋白质连接上核酸条形码,然后通过类似“转染”的方式将蛋白质-条形码复合物递送到细胞内[76]。同时,与临近连接方法(proximity ligation assay, PLA)[77]等其他PPI检测方法连用,实现在生理条件下动态检测PPI。
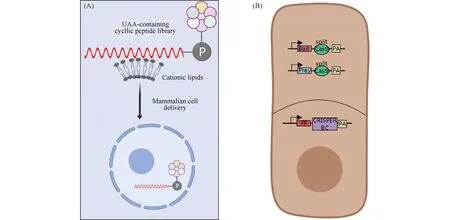
Fig.4 Potential applications of nucleic acid barcodes in PPI detection
其次,在条形码生成方面可以采用CRISPR-Cas9技术。目前,已有大量基于CRISPR-Cas9生成体内条形码的技术被相继开发[78],这些技术被用于动态记录细胞内的活动或实现细胞谱系追踪[79]。正如Fig.4B所示,如能采用CRISPR-Cas9编辑的动态条形码,则可显著增加实验通量,降低假阳性率。但有待解决的关键问题在于,通过何种方法可以将蛋白质相互作用与Cas9的活性联系起来。其中,基于分裂Cas9的蛋白质互补方法可能是一种解决方案[80],同时也借助DNA微阵列或IDPCR等技术,将sgRNA条形码与蛋白质连接起来,以提升PPI检测的效率和可靠性。
最后,是条形码连接技术的发展。目前的条形码连接技术,除上述提到的体内分子克隆的方法外,其余的2种方法均是将核酸条形码直接连接在目的蛋白质上,这就可能导致目的蛋白质的活性位点被掩盖,从而破坏蛋白质-蛋白质相互作用。最近提出的精准连接的概念[81]可能会解决这个问题。这一概念目前主要涉及2个技术:位点特异性原位生长(site-specific in situ growth, SIG)和内在无序多肽融合(intrinsically-disordered polypeptide fusion, IPF)。这2项技术的优点,首先是可以实现对蛋白质结合位点的精确控制,从而避免蛋白质活性位点被掩盖;其次,SIG的连接效率一般大于50%,而IPF的效率甚至可以达到100%,这有利于后续文库的纯化及质量评价。
同时,我们还期待未来技术的发展能带来更多的可能。例如,是否可以将单个细胞内的每个感兴趣的蛋白质从合成到降解的整个“生命周期”中都标记上条形码,每次与不同蛋白质的相互作用都被动态实时记录,以便更高的理解蛋白质相互作用在生命活动中的功能。总之,核酸条形码技术在大规模PPI检测方法中的应用仍处于蓬勃发展时期,在日后生命科学的各个研究领域会发挥更大作用。
