铁死亡在骨代谢中的调控机制
2024-04-23文皓楠海云翔董万涛宋志靖
文皓楠,王 凯,海云翔,董万涛,宋志靖,宋 敏,3
(1.甘肃中医药大学中医临床学院,甘肃 兰州 730000,2.甘肃中医药大学附属医院运动医学科,甘肃 兰州 730020;3.甘肃中医药大学附属医院老年骨科,甘肃 兰州 730020)
骨质疏松症(osteoporosis,OP)是一种由于各种因素导致的骨代谢平衡遭到破坏的慢性、全身性骨病,以骨密度降低和骨骼脆性增加为主要病理表现,目前,OP已经成为我国五十岁上人群的要健康问题,后期可导致脆性骨折,给家庭和社会带来了沉重的负担[1]。铁死亡(ferroptosis)是一种以铁依赖性脂质过氧化物积累为其特性的新型程序性细胞死亡模式,近年来受到医学领域的广泛关注。研究发现,铁死亡在调节OP发生、发展过程中发挥着关键作用。本文以“Ferroptosis”、“Bone metabolism”、“Osteoporosis”为关键词检索英文数据库PubMed、Cochrane、Embase、Web of Science,以“铁死亡”、“骨质疏松症”、“骨代谢”为关键词检索中文数据库万方(WANFANG DATA)、中国知网(CNKI)、维普(VIP),检索2012年至今的相关文献,对铁死亡在骨代谢领域取得的突破进行综述,探讨铁死亡的潜在机制以及其在不同骨稳态相关细胞中的调节作用,以期揭示OP的潜在治疗机制,拓展临床防治思路。
1 铁死亡特征及概述
铁死亡正式命名于2012年,stockwell等在前期研究的基础上,根据其铁依赖和氧化损伤的特性,将Erastin和RAS选择性致死化合物3(RAS-selective lethal 3,RSL3)诱导的非凋亡性细胞死亡(non-apoptotic cell death process)正式命名为铁死亡,其与其他细胞死亡的区分主要提现在以下方面:线粒体脊减少甚至消失是其主要的形态学特征,细胞内谷胱甘肽(glutathione,GSH)减少,谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)活性降低造成的细胞抗氧化能力减弱以及Fe2+在细胞内过度积累导致芬顿反应(Fenton Reaction),使活性氧(reactive oxygen species,ROS)和脂质过氧化物堆积发生过氧化反应是其主要的生化特征[2]。当Fe2+通过芬顿反应催化生成不稳定的羟自由基,参与多不饱和脂肪酸的脂质氧化,产生共轭二烯,进一步导致脂质过氧化产物4-羟基壬烯酸(4-hydroxynonenal,4-HNE)和丙二醛(malondialdehyd,MDA)等的生成,使得整个细胞膜的脆性增加,造成细胞功能减退,最终触发细胞铁死亡[3]。
2 铁死亡的调控机制
2.1 System xc-/GSH/GPX4/ROS轴调控氧化应激(oxidative stress,OS)是指体内氧化与抗氧化作用失衡的一种状态,当机体在遭受各种有害刺激时,体内高活性分子如ROS产生过多,可导致氧化系统和抗氧化系统失衡,最终导致组织损伤[4],而铁死亡因其铁依赖性的脂质过氧化特性,与氧化应激关系密切,一些学者甚至认为铁死亡是一种特殊的氧化应激损伤模式。System xc-是细胞内重要的抗氧化体系,为细胞表面的异二聚体-谷氨酸/胱氨酸转运体,其主要作用是实现胞内谷氨酸(glutamat,Glu)和胞外胱氨酸(cystin,Cys2)的互换,Cys2在谷氨酸半胱氨酸连接酶(glutamate-cysteine ligase,GCL)和谷胱甘肽合成酶(glutathione synthetase,GSS)的催化作用下合成GHS和GPX4,二者均是细胞内抗氧化系统的重要组成部分[2]。因此,System xc-的活性的抑制会降低GSH水平和GPX4活性,导致细胞抗氧化能力下降,从而诱导铁死亡的发生。
2.2 铁代谢——转运蛋白与“铁过载”调控铁是维持人体正常生命活动的重要微量元素,而铁代谢异常引发的“铁过载”是铁死亡发生的核心环节。在人体内,食物中Fe3+在肠道被还原为Fe2+后被小肠上皮细胞吸收,Fe2+在膜铁转运蛋白(ferroportin 1,FPN1)的作用下被运输至细胞外并被氧化为Fe3+,与转铁蛋白(transferrin,TF)结合形成TF-Fe3+复合物后进入血液循环,在到达靶细胞后,TF-Fe3+复合物与细胞膜上的转铁蛋白受体1(transferrin receptor 1,TFRC1)结合,通过胞吞的作用进入细胞内并被金属还原酶STEAP3还原为Fe2+[5]。胞内铁代谢平衡由Fe2+的储存和释放来维持,铁蛋白溶质载体家族40成员1(solute carrier family 40 member 1,SLC40A1)和铁蛋白重链1(ferritin heavy chain 1,FTH1)和轻链1(ferritin light chain 1,FTL1)以转运和储存的形式防止胞内铁过载,而核受体共激活因子4(nuclear receptor coactivator 4,NCOA4)在缺铁的条件下结合铁蛋白通过铁自噬防止胞内缺铁,若铁代谢紊乱,过量Fe2+在胞内聚集,发生芬顿反应,产生大量ROS,并与细胞膜上的多不饱和脂肪酸PUFA发生一系列过氧化反应造成细胞膜解体,最终引起铁死亡[6]。
2.3 脂代谢——多不饱和脂肪酸PUFAs过氧化调控脂质氧化物代谢失调也是造成铁死亡发生的核心因素。被酯化的多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)构成了生物膜的膜磷脂,是铁死亡中脂质过氧化物积累的关键底物,一些含有双烯丙基氢原子的物质,尤其是花生四烯酸(arachidonoyl,AA)和肾上腺素酸(adrenoyl,AdA),易与ROS发生反应并引起脂质过氧化,产生的脂质过氧化物可破坏磷脂双分子层的稳定性导致细胞膜的解体[7]。当长链脂酰辅酶A合成酶4(long train acyl CoA synthetases 4,ACSL4)催化游离的AA或AdA与辅酶A(coenzyme A,CoA)结合形成衍生物AA-CoA或AdA-CoA,被溶血卵磷脂酰基转移酶3(recombinant lysophosphatidylcholine acyltransferase 3,LPCAT3)酯化为多不饱和脂肪酸磷脂(poly-unsaturated fatty acids,PL-PUFA),并经过脂氧合酶(arachidonic acid lipoxygenase,ALOXs)或细胞色素P450氧化还原酶(cytochrome P450oxidoreductase,POR)氧化后,则会形成有害的脂质过氧化产物,诱导细胞铁死亡[8]。
2.4 NADPH/FSP1/CoQ10信号通路System xc-/GSH/GPX4/ROS轴被广泛认为是铁死亡的主要调控系统,但有研究发现,存在平行于该系统独立调控铁死亡的机制,NADPH/FSP1/CoQ10是首先被发现并最具代表性的调控信号通路。CoQ10是体内唯一能够自由合成的脂类抗氧化剂。2019年,Doll[9]等发现了一种线粒体凋亡诱导因子2(apoptosis inducing factor,mitochondria associated 2,AIFM2),因其过表达可显著抑制GPX4沉默诱导的铁死亡被命名为铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)。Kirill Bersuker[10]等发现,FSP1从线粒体转位到细胞膜后,通过还原性辅酶Ⅱ(nicotinamide adenine dinucleotide phosphate,NADPH)的酶促反应氧化还原CoQ10为CoQH2从而发挥其拮抗脂质过氧化物的功能,进而抑制细胞铁死亡,该研究为调控铁死亡拓展了新的路径。具体机制见Fig1。
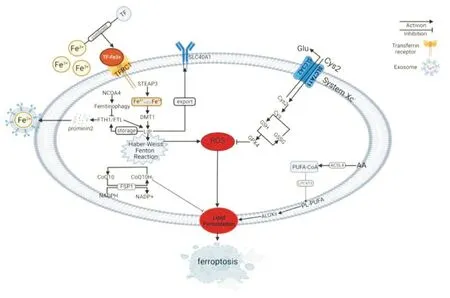
Fig1 Schematic diagram of ferroptosis regulation mechanism
3 铁死亡参与骨代谢的调控
OP的发病机制受到年龄、激素、内分泌等多方面因素影响,但其发病因素主是由于成骨细胞介导的骨形成与破骨细胞介导的骨破坏之间的平衡被打破,最终导致骨密度、骨质量下降和骨脆性增加。早期研究显示,铁过载及其产生的ROS积累可同时造成抑制骨形成和促进骨吸收的双向病理过程[11],近年来的大量研究证实,铁死亡对骨代谢的调控起到了关键作用。
3.1 铁死亡对骨髓间充质干细胞的影响骨髓间充质干细胞(bone mesenchymal stem cells,BMSCs)是存在于骨髓基质中具有多种分化潜能的细胞亚群,BMSCs的成骨、成脂分化失调是OP的重要病因。近年来一些研究已经证明,BMSCs铁死亡可降低其成骨分化能力从而诱发OP。Balogh等[12]证实铁过载可通过下调BMSCs成骨相关特异性基因RunX-2和成骨相关特异性基因和OCN的表达降低BMSCs的成骨分化,而过氧化氢和地塞米松(Dexamethasone,Dex)也被发现可诱导BMSCs铁死亡并降低其成骨分化能力[13]。调控BMSCs铁死亡也成为了OP治疗的新靶点,维生素E水解产物生育酚被发现可通过激活PI3K/AKT/mTOR信号通路抑制过氧化氢诱导的BMSCs铁死亡并促进成骨分化[14],可见通过抑制各种原因导致的BMSCs铁死亡以促进其成骨分化可能是治疗OP的潜在机制。
3.2 铁死亡对成骨细胞的影响成骨细胞(osteoblast,OB)由骨髓间充质干细胞分化而来,在骨基质合成,分泌和矿化中发挥重要作用,大量研究证实OB铁死亡会导致其分化和成骨能力下降,说明铁死亡是OP发病的重要机制之一。Luo等[15]发现在铁过载诱导铁死亡OB中发现促进骨形成的经典Wnt信号下调,而Wnt激动剂则能在不改变铁过载的前提下恢复OB分化。糖皮质激素及高糖均可诱导OB铁死亡,Lu等[16]研究发现10 μmol·L-1剂量的Dex即可通过下调GPX4和System xc-系统激活OB铁死亡。Ma等[17]发现高糖诱导可通过下调GPX4、升高ROS诱导MC3T3-E1铁死亡。这些都表明,通过各种途径抑制OB铁死亡以促进骨形成具有广阔的运用前景。
3.3 铁死亡对破骨细胞的影响破骨细胞(osteoclast,OC)是由单核巨噬细胞系或BMSCs经NF-κB配体受体激活剂(RANKL)的诱导形成多核大细胞,执行骨吸收功能。研究显示OC铁死亡能降低其活性导致骨吸收减少[18],而与之矛盾的是,大量研究表明引发铁死亡通路的铁过载和过量ROS生成均可促进OC生成,铁过载可促进骨细胞凋亡并增加RANKL诱导的破骨细胞分化,RANKL可增强铁自噬作用导致胞内铁离子增加,而随着铁离子增加通过芬顿反应产生的ROS可促进破骨细胞分化[19],上述机制以RANKL为轴交互作用,形成“铁过载-RANKL增加-铁蛋白自噬-加重铁过载-骨吸收增加”的恶性循环,该矛盾的产生可能与OC分化过程中独特的嗜铁性以及ROS在OC分化中充当细胞间途径中有效的多功能信号分子有关,其机制有待进一步探究。而通过不同途径打破该循环成为调控破骨细胞分化治疗OP的靶点。青蒿琥酯可通过p62/Nrf2信号通路抑制ROS的产生抑制OC生成[20],使用铁螯合剂去铁氧胺(DFO)可在体外通过MAPK通路抑制破骨细胞生成[21]。此外,诱导OC铁死亡也成为治疗OP的靶点,HIF-1α特异性抑制剂2-甲氧基雌二醇(2-methoxyestradiol,2ME2)可通过增加铁自噬诱导OC铁死亡以改善卵巢切术后(Ovariectomy,OVX)小鼠OP[22],双膦酸盐作为治疗OP的常用药物,在近期被发现可通过触发FBXO9介导的p53泛素化和降解来诱导破骨细胞铁死亡以改善OP的新机制[18]。总之,调节破骨细胞的铁死亡可能是治疗骨质疏松症的一个选择。
综上,铁死亡在骨代谢中因其作用的细胞不同而发挥着相应的生物学作用,BMSCs铁死亡可抑制其成骨分化,OB铁死亡可降低骨形成,OC铁死亡则抑制骨吸收(如Fig2所示)。深入研究铁死亡在骨稳态中与OB、OC耦联的机制,从多靶点,多环节抑制铁死亡在OP发病过程中的作用,是以铁死亡作为切入点防治OP的前景所在。

Fig2 Schematic diagram of regulatory mechanism of ferroptosis on bone metabolism
4 铁死亡在骨质疏松治疗中的运用
4.1 铁死亡与糖尿病性骨质疏松糖尿病性骨质疏松(diabetic osteoporosis,DOP)是常见的继发性骨质疏松之一,近年来,大量的研究证实了铁死亡是DOP发病的重要环节,高糖环境被证实可通过诱导BMSCs铁死亡并抑制其成骨分化[23],靶向抑制铁死亡逐渐成为DOP治疗的研究热点。Ma等[17]发现,褪黑素可通过激活Nrf2/HO-1通路降低高糖诱导的MC3T3-E1细胞铁死亡水平,并增强其成骨能力。线粒体铁蛋白(ferritin in mitochondria,FtMt)是一种存在于线粒体中,具有储存铁离子功能的蛋白质。Wang等[24]发现,过表达FtMt可减少高糖状态下OB的铁死亡,而沉默FtMt可激活ROS/PINK1/Parkin信号通路激活线粒体自噬并释放铁离子,促进成骨细胞铁死亡,提示FtMt可能是治疗DOP的潜在靶点。Zhang等[25]通过研究发现,由多不饱和脂肪酸产生的内源性促分解脂质介质(maresin1,MaR1)在体内和体外均能激活Nrf2通路,抑制高糖高脂诱导的OB铁死亡。通过铁死亡途径干预糖尿病导致的骨形成减弱是目前新兴的DOP治疗途径,铁死亡对DOP调控的分子机制以及铁死亡如何影响DOP的发病仍有待进一步研究。
4.2 铁死亡与激素性骨质疏松糖皮质激素(glucocorticoid,GC)作为抗炎和免疫抑制剂,在临床上运用广泛。大量、长期使用GC对骨骼和肌肉组织造成不可逆的损伤,从而导致糖皮质激素性骨质疏松症(glucocorticoid-induced osteoporosis,GIOP)。研究显示,GC诱导的OB铁死亡所导致的骨形成减少是导致GIOP的重要机制之一[16]。大量研究者基于该机制拓展了GIOP的治疗途径,Sun等[26]在研究中发现,Dex通过抑制MC3t3-E1细胞SLC7a11/GPX4的表达,降低细胞内GSH活性并增加ROS含量诱发铁死亡,而敲减SLC7a11的上游基因p53可逆转Dex对MC3t3-E1细胞中SLC7a11/GPX4表达的抑制并防止铁死亡的产生,该实验从基因层面拓展了GIOP的治疗思路。Lu等[27]研究中证实,骨髓来源的内皮祖细胞中提取的胞外囊泡(bone marrow EPCs,BM-EPCs)可通过上调SLC3A2,SLC7A11和GPX4逆转Dex导致的骨细胞铁死亡,该实验证实了通过干预骨细胞铁死亡以治疗GIOP是可行的方向。综上,通过抑制GC造成的脂质过氧化和铁过载造成的BMSCs及OB铁死亡,是目前通过干预铁死亡治疗GIOP的主要手段,明确节点通路和关键靶点并从不同途径抑制GC对骨代谢的影响,是GIOP的治疗方向之一。
4.3 铁死亡与原发性骨质疏松PMOP是由绝经后妇女雌激素缺乏引起的原发性骨质疏松,其主要病因是由于雌激素缺乏导致骨吸收增强,骨形成减弱,最终导致骨量减少和骨脆性增加,早期研究显示,绝经后妇女血清游离铁含量高于正常人群,铁代谢紊乱可能参与了PMOP的发病过程[28]。Ni等[22]证实2ME2可通过增强骨髓源巨噬细胞(bone marrow-derived macrophages,BMDMs)的铁自噬诱导铁死亡,进而降低OVX小鼠骨吸收以改善OP,表明以HIF-1α和铁蛋白为靶点诱导破骨细胞的铁死亡可能是治疗PMOP新方法。
虽然众多研究已证明绝经后的低雌激素水平可能与铁死亡相关,但目前仍无证据说明二者间的直接关系,其影响机制有待进一步研究。
老年性骨质疏松症(senile osteoporosis,SOP)是一种增龄相关的全身性骨病,研究显示,在衰老过程中铁代谢逐渐失衡,非血红素铁积累造成细胞内环境稳态的破坏是一系列衰老相关疾病的病因[29]。Xu[30]等发现,D-半乳糖(D-gal)诱导的衰老小鼠SOP可能与铁死亡有关,而维生素D受体激活剂1, 25(OH)D可通过Nrf2/GPX4通路抑制SOP小鼠体内铁死亡水平并增加骨形成,该实验初步验证了SOP与铁死亡的相关性,并说明通过干预维生素D代谢调控铁死亡可能是治疗SOP的新方向。总之,调控骨代谢相关细胞铁死亡可能是治疗OP的新策略所在。具体机制见Tab1。

Tab1 Mechanism of ferroptosis in preventing and treating osteoporosis
5 总结与展望
铁死亡作为一种新型的程序性细胞死亡,其在OP发病过程的作用机制被逐渐揭示,已成为防治OP的研究热点。内分泌、激素、绝经及高龄等因素通过铁死亡影响骨代谢的机制已被初步明确,通过抑制上述原因造成的骨形成相关细胞铁死亡或激活骨吸收相关细胞铁死亡成为目前通过铁死亡途径防治OP的主要思路。调控骨代谢相关细胞铁死亡以恢复骨吸收和骨形成之间的平衡具有广阔的挖掘空间。但铁死亡与骨代谢的研究仍然处于起步阶段,仍存在一些问题以亟解决:①目前对铁死亡对OP的调控研究多集中于铁过载和脂质过氧化两种机制,随着铁死亡的新路径不断被揭示,是否存在其他调控机制以及触发因素仍待探索。②在骨稳态的调节中,铁死亡与其他形式的程序性死亡如焦亡、自噬之间的互作关系仍未明确,需要进一步深入研究。③受制于动物模型的因素,对于老年性OP与铁死亡的研究仍然较少,需要建立科学的动物模型以明确铁死亡与原发性OP的关系;④铁死亡如何通过BMSCs、OB和OC之间的耦联及对骨稳态进行精密的调控机制仍需深入挖掘;⑤中药单体、复方及针刺治疗OP具有多靶点、多途径、多效应的调控特色,可通过进一步研究来明确中医药介导铁死亡治疗OP的途径,拓展中医药治疗OP的新靶点。⑥炎症与免疫原性作为影响骨代谢的重要因素,与铁死亡的关系在其他疾病中已经被揭示,然而目前尚未有研究说明二者是否通过铁死亡与骨代谢的调控相关。⑦选择性的在体内控制骨代谢相关铁死亡的机制和方法仍然难以捉摸,在特定组织、细胞和或疾病背景下选择性激活或抑制铁死亡是将目前的研究转化为临床应用的关键。
