1例c.1117C>T/c.7288-9T>G复合杂合突变所致血管性血友病家系的发病机制分析
2024-04-22谭忠州陆遥苗林子李园园朱梓静宋一楠龚岩屈晨雪
谭忠州,陆遥,苗林子,李园园,朱梓静,宋一楠,龚岩,屈晨雪
(北京大学第一医院检验科,北京100034)
血管性血友病(von Willebrand disease, vWD)是一种以血管性血友病因子(von Willebrand factor, vWF)数量或质量缺陷为特征的常见遗传性出血性疾病。vWF是由内皮细胞和巨核细胞合成的多聚体糖蛋白[1-2]。当血管内皮细胞损伤时,在高血流剪切力作用下vWF可介导血小板与内皮下胶原的黏附,此外vWF还可作为血浆FⅧ的载体,保护其不被血浆中的丝氨酸蛋白酶(如蛋白酶C)水解,延长其半衰期[3]。因此vWF功能缺陷患者主要临床表现为自发性皮肤黏膜出血、拔牙或术后过度出血。
根据国际血栓与止血委员会(International Society on Thrombosis and Haemostasis, ISTH)的建议可将vWD分为1型(vWF数量部分缺乏)、2型(vWF质量或数量缺乏)和3型(vWF数量完全缺乏)。其中2型vWD又可进一步分为2A型(vWF高分子多聚体减少或缺乏)、2B型(vWF和血小板亲和力增加)、2M型(vWF和血小板或胶原结合能力减低)和2N型(vWF与FⅧ结合能力减低)[4]。vWD诊断是一个复杂的过程,需要收集患者及家系的出血史并进行多种检测,包括止血筛查试验、vWD诊断实验和分型试验。
本研究将对临床1例可疑vWD患者及其家系进行诊断,并采用全外显子组测序和生物信息学等方法探讨该家系的发病机制,为探寻更好的临床诊断、治疗路径和方案提供更多理论依据。
1 材料和方法
1.1对象 先证者(Ⅲ1),女,13岁,自3岁起易发生自发性鼻黏膜出血,每天1~2次,持续约30分钟,不易止血。先证者的母亲(Ⅱ4)磕碰后皮肤易形成瘀斑,哥哥(Ⅲ2)和父亲(Ⅱ3)无明显出血表现。父亲家族无手术史、家族史,先证者的外婆(Ⅰ3)易发生牙龈出血(见图1)。
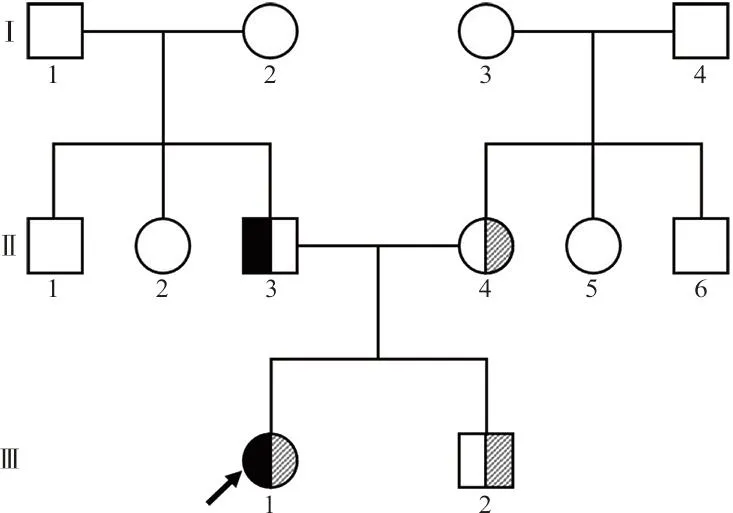
注:,vWF:c.7288-9T>G杂合突变;,vWF:c.1117C>T杂合突变;,vWF:c.1117C>T杂合突变;,vWF:c.1117C>T和c.7288-9T>G杂合突变;□,男性;○,女性;,先证者。
本研究涉及1个患者家系共4人,通过北京大学第一医院伦理委员会审核[批准文号:(2019)科研第(47)号]。
1.2方法
1.2.1标本采集与处理 收集先证者及其家系成员0.109 mol/L枸橼酸钠抗凝血、 1.8 mg/mL乙二胺四乙酸二钾(EDTA-K2)抗凝血及惰性分离胶促凝血。其中EDTA-K2抗凝血用于全血细胞分析、镜下血小板形态检查及外周血基因组DNA提取。惰性分离胶促凝血用于免疫球蛋白IgA、IgG、IgM及生化全项检查。枸橼酸钠抗凝血用于活化部分凝血酶时间(activated partial thromboplastin time APTT)、凝血酶原时间(prothrombin time, PT)、纤维蛋白原定量(fibrinogen concentration, Fib-C)、凝血酶时间(thrombin time,TT)、D-二聚体及纤维蛋白(原)降解产物(fibrin or fibrinogen degradation products,FDPs)分析以及vWF活性(vWF activity, vWF:Ac)、vWF抗原(vWF:Ag)、凝血因子Ⅷ活性(factor Ⅷ activity, FⅧ:C)、vWF瑞斯托霉素辅因子试验(vWF ristocetin co-factor,vWF:RCo)、瑞斯托霉素诱导的血小板凝集试验(restocetin-induced platelet aggragation,RIPA)、vWF胶原结合试验(vWF collagen binding assays, vWF:CB)。
1.2.2实验室检查 用BC-6800Plus血细胞分析仪(深圳迈瑞公司)进行全血细胞分析;ACL-TOP700全自动血凝分析仪(西班牙沃芬公司)进行APTT、PT、Fib-C、TT、FDP、vWF:Ac及vWF:Ag检测;用CS2000i分析仪(日本希森美康公司)检测FⅧ:C;用日立H7600-4全自动生化分析仪进行生化全项检查;采用IMAGE800特种蛋白分析仪(美国贝克曼库尔特公司)进行免疫球蛋白IgG、IgA和IgM检测;采用LBY-NJ14血小板聚集仪(中国泰利康信公司)进行RIPA、vWF:RCo试验,瑞斯托霉素购自美国Chrono-log公司;vWF:CB采用ZYMUTESTTMvWF:CBA试剂盒(法国HYPHEN BioMed公司)进行检测。
1.2.3全血基因组DNA提取 采用Lab-Aid 824核酸提取Midi试剂盒(厦门致善公司)提取先证者及其家系成员的EDTA-K2抗凝全血基因组DNA。
1.2.4基因检测及分析 提取的先证者及其家系成员的外周血基因组DNA进行全外显子组测序分析并对发现的vWF基因突变位点采用一代测序方法进行验证。采用MutationTaster、CADD、SIFT和PolyPhen-2以及NetGene2等生物信息分析工具预测基因突变位点的致病性。
2 结果
2.1实验检查结果
2.1.1常规检查 血常规、生化常规、免疫球蛋白(IgA、M和G)等常规实验检查及镜下血小板形态分析,先证者及家系成员未见明显异常。除先证者APTT 37.8 s为轻度延长外,家系其他成员(父亲、母亲及哥哥)结果均正常,Fib-C、PT、TT及FDP检测先证者及家系成员结果均正常,见表1。

表1 先证者及其家系常规凝血结果
2.1.2vWD相关实验检查 FⅧ:C先证者在参考区间的偏低水平,家系其他成员(父亲、母亲及哥哥)均正常。先证者vWF:Ag为27.6%,明显减低,哥哥为48.6%,轻度减低,父亲和母亲均正常。与正常对照相比,先证者及家系成员的vWF:Ac、vWF:CB均有不同程度减低,其中先证者(14%、10.37%)减低最为显著。先证者、哥哥及父亲vWF:RCo均有不同程度减低。1.25 mg/mL RIPA试验显示先证者(12%)及哥哥(45%)均减低,且1.0 mg/mL瑞斯托霉素诱导下RIPA试验先证者及其家系成员均减低,见表2。

表2 先证者及其家系vWF相关检查结果
2.2基因突变分析结果
2.2.1DNA测序结果 全外显子组测序发现先证者存在vWF基因编码区(转录本号:NM_000552)第7 288号核苷酸前内含子(42号内含子)中倒数第9位氨基酸由T突变为G(c.7288-9T>G)、第1 117号核苷酸由C突变为T(c.1117C>T)的复合杂合突变,父亲vWF基因存在c.7288-9T>G杂合突变,母亲和哥哥vWF基因均存在c.1117C>T杂合突变(表3、图2、图3)。

表3 先证者及家系成员的临床特征与基因分析结果

注:A为父亲,野生型;B为母亲,杂合突变型;C为先证者,杂合突变型;D为哥哥,杂合突变型。

注:A为父亲,杂合突变型;B为母亲,野生型;C为先证者,杂合突变型;D为哥哥,野生型。
2.2.2生物信息学分析结果 采用NetGene2、NNSplice、FSPLICE和MaxEntScan对vWF基因c.7288-9T>G突变进行剪切位点预测分析,突变前(后)预测分数分别为0.81(0.19)、0.88(0.54)、剪切位点正常(剪切位点破坏)和-4.17(-11.12),见表4。

表4 vWF基因突变位点致病性预测
3 讨论
本研究报道了1例遗传性vWD家系。先证者为女性,表现为皮肤黏膜出血,其血小板计数正常,但APTT轻度延长,FⅧ:C接近参考区间下限,vWF:Ag明显减低,因此,考虑该患者可能为vWD。为了明确诊断和分型,本研究进一步检测了vWF:Ag、vWF:Ac、vWF:RCo、RIPA试验以及vWF:CB试验。vWF:Ag反映血浆vWF多聚体的抗原性。vWF:RCo和vWF:Ac试验直接反映vWF多聚体A1结构域的GpⅠbα结合位点活性。RIPA试验反映了vWF与自身血小板的结合能力。vWF:CB试验反映vWF与胶原结合能力,对vWF高分子多聚体缺失敏感,当vWF高分子多聚体缺失时,vWF:CB具有不同程度减低。
先证者vWF:Ac、vWF:RCo及vWF:CB明显减低,1.25 mg/mL及1.0 mg/mL瑞斯托霉素诱导下RIPA试验也明显减低,且vWF:CB/vWF:Ag小于0.6[5-7],这提示vWF高分子量多聚体减少。根据最新vWD诊断指南[8-9],该患者最可能是2型vWD。通过全基因组测序并未发现GPIbα基因相关突变且低剂量瑞斯托霉素诱导下RIPA试验并无明显聚集,故排除2B型vWD,最终先证者(Ⅲ1) 诊断为2A型vWD。哥哥(Ⅲ2)的FⅧ:C正常,vWF:Ag、vWF:Ac、vWF:RCo及vWF:CB轻度减低,1.25 mg/mL及1.0 mg/mL瑞斯托霉素诱导下RIPA活性减低。根据英国血友病中心医师组织(UKHCDO)建议vWF:Ag小于50%,大于30%且伴随异常出血症状的患者可诊断为“低vWF”[10],但哥哥(Ⅲ2)不表现明显出血症状,不符合“低vWF”诊断。母亲(Ⅱ4)的FⅧ:C、vWF:Ag及vWF:RCo正常,vWF:Ac、vWF:CB及1.0 mg/mL瑞斯托霉素诱导的RIPA活性减低。父亲(Ⅱ3)的FⅧ:C、vWF:Ag、vWF:Ac及1.25 mg/mL瑞斯托霉素诱导下RIPA活性正常,而vWF:RCo、vWF:CB及1.0 mg/mL瑞斯托霉素诱导下RIPA活性相比正常对照均有不同程度减低。
为进一步探讨先证者(Ⅲ1)的发病机制,对该家系4位成员进行全外显子测序分析,结果发现先证者(Ⅲ1)以及其母亲(Ⅱ4)、哥哥(Ⅲ2) 均存在vWF基因编码区第10号外显子c.1117C>T(rs62643625)杂合突变,检索dbSNP数据库发现,突变c.1117C>T在人群中的发生频率极低,不属于多态性变化。该突变为无义突变,导致vWF前肽第373号氨基酸(精氨酸)的密码子变为终止密码子,从而使肽链合成终止在vWF前肽的D1与D2结构域之间,这就导致合成的vWF分子无功能结构域,因此携带有该突变的患者FⅧ:C、vWF:Ag、vWF:Ac和vWF:CB等结果不同程度减低。文献报道携带有该纯合突变患者或复合杂合突变患者表现出严重的出血症状,vWF:Ag小于1%、FⅧ:C小于5%,诊断为3型vWD[11-13]。基因测序结果还显示先证者(Ⅲ1)及其父亲(Ⅱ3)均存在vWF基因编码区第42号内含子c.7288-9T>G(rs1348526788)杂合突变。该突变为内含子剪切突变,其致病性尚未见文献报道。进一步分析发现,先证者(Ⅲ1)表现出明显出血症状,但携带vWF基因c.1117C>T杂合突变的母亲(Ⅱ4)和哥哥(Ⅲ2)无明显出血表现,因此推测先证者(Ⅲ1)的发病除来自母亲(Ⅱ4)遗传的c.1117C>T突变外,遗传自父亲(Ⅱ3)的c.7288-9T>G突变可能也发挥作用,两种基因突变共同作用导致先证者(Ⅲ1)2A型vWD表型。通过生物信息软件预测c.7288-9T>G突变的致病性及携带有这一杂合突变的父亲(Ⅱ3)表型一定程度验证了推测。
本研究通过一系列实验室检查诊断了1例血管性血友病家系,对该患者的后续预防及临床治疗非常重要,同时也对vWD实验诊断具有启示作用。但在测量血小板依赖的vWF活性时,本研究采用的传统的vWF:RCo方法相对新的vWF:GPⅠbM和vWF:GPⅠbR方法变异较大,故为提高诊断的准确性,建议应采用新的vWF:GPⅠbM或vWF:GPⅠbR方法测量血小板依赖的vWF活性[8-9]。此外,本例家系检测到的新发突变vWF基因内含子c.7288-9T>G突变,仅仅通过生物信息学预测可能具有致病作用,故还需进一步进行体外实验来验证该突变的致病性。
