后纵韧带骨化症患者基因变异及表型分析
2024-04-19陈欣张立周非非赵衍斌刁垠泽潘胜发王少波张凤山孙宇
陈欣,张立,周非非,赵衍斌,刁垠泽,潘胜发,王少波,张凤山,孙宇
后纵韧带骨化症(ossification of the posterior longitudinal ligament, OPLL)是以后纵韧带异常增生并骨化为特点的疾病,可压迫神经根导致严重的神经功能障碍甚至高位截瘫。中国人颈椎OPLL 和胸椎OPLL 的患病率分别为4.1%和2.25%[1]。流行病学和家系研究均表明遗传因素参与OPLL 的发生与发展[2]。陈振等[3]也发现颈椎OPLL 家族聚集性人群OPLL 发生率较普通人群明显升高,提示遗传因素在颈椎OPLL发生机制中占重要地位[3]。虽然利用全基因组关联分析(genome wide association study,GWAS)[4]、连锁分析和病例-对照相关性研究[5]已发现多个与OPLL 相关的基因/基因座,但其中仅有Enpp1基因纯合缺陷小鼠[6-7]出现了类似于人类OPLL 的表型。有报道ENPP1基因变异可导致全身性婴儿动脉钙化(OMIM:#208000)和低磷血症性佝偻病(OMIM:#613312),其中个别病例伴有OPLL 的发生,呈常染色体隐性遗传[8]。然而,最近有报道ENPP1单等位基因变异也可导致OPLL 的发生[9]。本研究通过对OPLL 患者进行全外显子组测序(whole exome sequencing, WES),明确其可能的致病原因。
1 资料与方法
1.1 一般资料
纳入标准:①经临床症状、体格检查及脊柱正侧位X 线片诊断为OPLL;②签署知情同意书。排除标准:①正在参加其他干预性临床试验者;②有精神障碍或认知障碍者;③内分泌系统疾病患者;④心肺疾病患者;⑤神经系统疾病患者;⑥严重的肝肾疾病、肿瘤患者。
2017年1月至2019年12月北京大学第三医院收治OPLL 患者125 例,依据上述纳入与排除标准选取93 例患者进行WES。其中男56 例,女37 例,年龄33~75岁,平均(52.1±11.0)岁。
本研究经北京大学第三医院伦理委员会审批通过(IRB00006761-2015029),所有患者均签署知情同意书。
1.2 基因组DNA提取
取患者EDTA 抗凝外周静脉血2 mL,采用QIAamp 全血DNA 提取试剂盒(德国Qiagen 公司)提取基因组DNA。
1.3 WES
将基因组DNA 打断、纯化,使用Agilent SureSelect Human All Exome V6 试剂盒(加拿大Agilent Technologies公司)进行全基因组外显子捕获,应用Illumina Hiseq X Ten测序仪(美国Illumina公司)进行读长为150 bp的双端高通量测序,测序得到的数据经过质量控制,运用序列比对软件Burrows-Wheeler Aligner(BWA)[10]与GRCh37/hg19人类参考基因组进行比对,利用ANNOVAR[11]进行变异位点注释。
1.4 生物信息学分析
利用千人基因组、dbSNP、gnomAD和ExAC数据库分析变异频率,如果变异为杂合,滤掉次等位基因频率>0.005 的普遍变异,如变异为纯合或同一基因中有2个或以上杂合变异,滤掉次等位基因频率>0.02的普遍变异。利用预测软件SIFT、Polyphen-2、Mutation Taster、CADD 等进行变异的有害性预测。筛选出剪接位点变异、移码变异、无义变异及至少2个预测软件预测为有害的错义变异。选择GWAS、连锁分析和病例-对照相关性研究得到的OPLL 相关基因,从过滤后的变异中筛选患者中出现的OPLL 相关基因变异。查阅在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man, OMIM)、ClinVar、人类基因突变数据库(Human Gene Mutation Database,HGMD)是否收录该变异,利用UCSC(http://genome.ucsc.edu)对变异进行同源序列保守性分析,利用InterPro(https://www.ebi.ac.uk/interpro/)进行变异位点结构域分析,利用UCSF chimera[12]进行蛋白三维结构分析,综合判断变异的致病性。
1.5 ENPP1基因变异患者临床表型分析
根据影像学表现对OPLL 进行分型[13]。手术前后采用日本骨科协会(Japanese Orthopedic Association, JOA)评分评估患者神经功能。对患者进行全身体检,明确有无佝偻病表型。血清生化检查测定血清钙、磷、碱性磷酸酶水平。
2 结果
2.1 WES结果
选择GWAS 研究得到的基因(HAO1、RSPO2、EIF3E、EMC2、CCDC91、EIF3H、CDC5L、SUPT3H)[4]及连锁分析和病例-对照相关性研究得到的基因(TLR5、RXRB、COL11A2、RUNX2、IL1B、ENPP1、ESR1、IL15RA、BMP9、VDR、BMP4、TGFB3、TGFB1、BMP2、COL6A1)[5]作为OPLL相关基因。本研究通过生物信息学分析,在2 例OPLL 患者中发现了ENPP1基因变异。其中,在患者1 中发现了ENPP1基因的1 个杂合错义变异c.T802C(p.Tyr268His),在患者2中发现了2 个ENPP1基因的杂合错义变异c.T253C(p.Cys85Arg)和c.T802C(p.Tyr268His),其中ENPP1c.T802C(p.Tyr268His)在2 例患者中均被发现。患者1和患者2均未发现其他OPLL相关基因的罕见变异。

图1 ENPP1基因变异位点保守性分析
2.2 变异致病性分析
ENPP1错义变异c.T253C(p.Cys85Arg)在4 个人群数据库(千人基因组、dbSNP、gnomAD、ExAC)中均未被收录。3 个疾病数据库(OMIM、ClinVar、HGMD)均未收录该变异。多种蛋白预测软件SIFT、Polyphen-2、Mutation Taster 均预测该变异有害或可能有害,CADD评分为25.9分。
ENPP1错义变异c.T802C(p.Tyr268His)在千人基因组频率为0.000 2,gnomAD 数据库人群频率为8.57×10-5,ExAC 数据库人群频率为7.9×10-5,dbSNP数据库收录该变异(rs17847050)。OMIM、ClinVar、HGMD 数据库均未收录该变异。多种蛋白预测软件SIFT、Polyphen-2、Mutation Taster 均预测该变异有害,CADD评分为25.6分。
ENPP1基因c.T253C(p.Cys85Arg)和c.T802C(p.Tyr268His)变异位点在不同物种中均具有高度保守性(图1)。
ENPP1基因在结构上分为N-末端胞质结构域、跨膜结构域、串联生长介素B 样结构域1、串联生长介素B 样结构域2、磷酸二酯酶催化结构域、L1接头、L2接头、C-末端核酸酶样结构域[14]。ENPP1c.T253C(p.Cys85Arg)位于跨膜结构域,c.T802C(p.Tyr268His)位于磷酸二酯酶催化结构域(图2)。利用UCSF chimera 软件分析ENPP1 蛋白三维结构(PDB ID:6WFJ),评估氢键形成及氨基酸之间的距离,发现268 位的酪氨酸(Tyr268)残基可以与His500、Glu565、Arg888、Asp891 之间形成极性相互作用。赖氨酸残基转化为组氨酸(His268)使其与His500、Glu565、Arg888、Asp891 的极性相互作用消失,与Glu270之间形成新的极性相互作用(图3)。
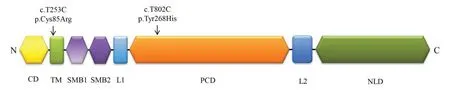
图2 OPLL蛋白结构域示意图

图3 ENPP1蛋白三维结构示意图

图4 患者1影像学表现

图5 患者2影像学表现
2.3 2例ENPP1基因变异患者临床表型与实验室检查
患者1表现为颈椎OPLL、胸椎OPLL、胸椎黄韧带骨化症(图4),颈椎OPLL分型为节段型(C3~C7),无身材矮小和佝偻病表型,身高181 cm,血清钙、磷和碱性磷酸酶水平正常,分别为2.23 mmol/L、0.98 mmol/L、61 U/L,颈椎JOA 评分8 分。患者无高血压、糖尿病(表1)。
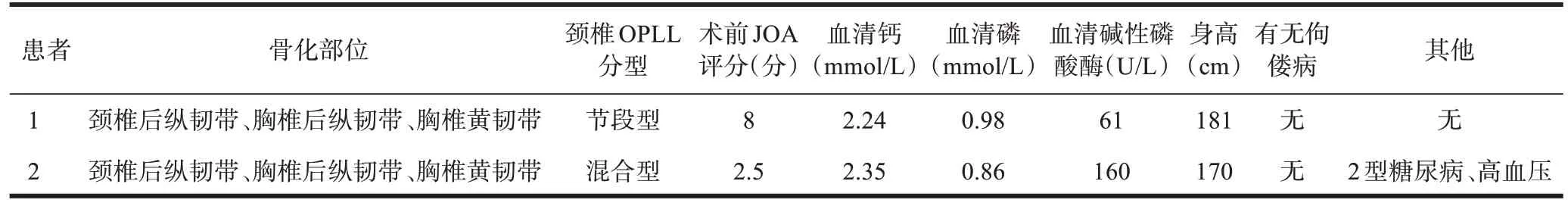
表1 OPLL患者临床表型分析
患者2 表现为颈椎OPLL、胸椎OPLL、胸椎黄韧带骨化症(图5),OPLL分型为混合型(连续型C2~C5,局灶型C6、C7,连续型C7~T2),无身材矮小和佝偻病表型,身高170 cm,血清钙水平正常,为2.35 mmol/L,血清磷水平在正常低限,为0.86 mmol/L,血清碱性磷酸酶水平升高,为160 U/L。颈椎JOA评分2.5分。患者伴有2型糖尿病和高血压(表1)。
3 讨论
OPLL 的发病与遗传和环境因素有关。一项GWAS 研究表明多个促进骨化的基因如HAO1、RSPO2和CCDC91等与OPLL 发生、发展有关[4]。另外,连锁分析和病例-对照相关性研究也发现了一些与OPLL 相关的基因/基因座,包括编码核苷酸焦磷酸酶/磷酸二酯酶1 的基因(ENPP1)、编码细胞外基质蛋白的基因(COL11A2、COL6A1)、编码骨形态发生蛋白的基因(BMP2、BMP4、BMP9),以及编码TGF-β 信号通路蛋白的基因(TGFB1、TGFB3)等[5]。陈振等[3]对家族性OPLL 的研究发现,家族性OPLL在同辈中的发生率为50.0%(13/26),符合常染色体显性单基因遗传病在患者同胞中的发病率,表明一部分OPLL的发病可能是由单个基因变异引起。
ENPP1基因编码核苷酸焦磷酸酶/磷酸二酯酶1(ectonucleotide pyrophosphatase/phosphodiesterase 1),定位于染色体6q23.2 上,由编码925 个氨基酸的25个外显子组成。ENPP1 是一种膜结合糖蛋白,通过水解细胞外三磷酸核苷酸(nucleotide triphosphates,NTP)产生无机焦磷酸(pyrophosphate, PPi)。正常水平的PPi 可以防止软组织和血管钙化[15],ENPP1 功能障碍导致PPi 产生减少,从而导致异位钙化的发生。具有Enpp1基因p.Gly568X 纯合变异的ttw小鼠表现出脊柱韧带的异位骨化,这种表型类似于人类OPLL[6]。然而,ENPP1基因变异导致OPLL 的报道较为少见[8-9,16-17]。
本研究通过WES 和生物信息学分析,在2 例OPLL 患者中发现了ENPP1基因的2 个错义变异,其中患者1 携带c.T802C(p.Tyr268His)杂合变异,患者2 携带c.T253C(p.Cys85Arg)杂合变异和c.T802C(p.Tyr268His)杂合变异。由于没有患者父母的血液样本,无法验证患者2中发现的2个ENPP1变异是否为复合杂合变异。c.T253C(p.Cys85Arg)和c.T802C(p.Tyr268His)变异位点在不同物种中均具有高度保守性,多种蛋白预测软件均预测变异有害和可能有害。
2 例患者均携带的变异c.T802C(p.Tyr268His)位于磷酸二酯酶催化结构域,该结构域是介导ENPP1催化活性的结构域[18]。ENPP1 包括8 个结构域,目前已报道的140 个ENPP1基因变异大多数集中在磷酸二酯酶催化结构域和核酸酶样结构域,分别占所有变异的49.3%和27.1%[19]。蛋白三维结构分析发现该变异可能影响其与周围氨基酸之间的极性相互作用,从而影响酶活性,导致OPLL 的发生。Ferreira等[8]曾报道1 例患者携带c.T802C(p.Tyr268His)相同位置不同氨基酸改变c.803A>G(p.Tyr268Cys)。该患者携带ENPP1c.803A>G(p.Tyr268Cys)和c.2596G>A(p.Glu866Lys)的复合杂合变异,出现了全身性婴儿动脉钙化、低磷血症性佝偻病和OPLL(C2~C3 和C3~C4),并伴有肾病、糖尿病、高血压和高脂血症。这些发现支持ENPP1c.T802C(p.Tyr268His)可能是2例患者出现椎旁韧带骨化症的原因。ENPP1c.T802C(p.Tyr268His)在gnomAD 和ExAC 数据库的频率分别为8.57×10-5和7.9×10-5,然而在gnomAD 东亚人群和ExAC 东亚人群数据库的频率分别为0.001 和0.000 7,通过查询中国人群全基因测序“女娲”数据资源[20],发现ENPP1c.T802C(p.Tyr268His)在中国人群中的频率为0.001 8(11/5 998),显著高于其他人种,说明该变异有可能是中国人群OPLL高发的原因之一。
除了c.T802C(p.Tyr268His),本研究还发现患者2携带ENPP1的另一个杂合变异 c.T253C(p.Cys85Arg)。c.T253C(p.Cys85Arg)位于跨膜结构域。目前仅有c.241G>T(p.Val81Leu)、c.272T>C(p.Leu91Pro)和c.275G>A(p.Gly92Asp)这3个错义变异被报道位于跨膜结构域[19]。其中c.241G>T(p.Val81Leu)纯合变异可导致全身性婴儿动脉钙化,而患者携带杂合变异的父母未发病。功能研究发现ENPP1 c.241G>T可引起异常剪接,导致2号外显子跳跃[21]。然而ENPP1c.T253C(p.Cys85Arg)通过何种机制致病仍需实验验证。
ENPP1基因纯合或复合杂合变异可导致全身性婴儿动脉钙化和低磷血症性佝偻病。全身性婴儿动脉硬化表现为大中型动脉的内弹性层钙化和狭窄,严重时与心血管衰竭和早期死亡相关,在胚胎时期或出生6个月内死亡率约为55%,血管外钙化也常见于幸存者的关节、脊柱和内脏组织[22]。低磷血症性佝偻病表现为身材矮小、骨骼畸形、低磷酸盐血症、碱性磷酸酶水平升高和成纤维细胞生长因子23(fibroblast growth factor 23, FGF23)水平升高或正常高限[23]。此外,ENPP1基因变异可导致Cole病(OMIM:#615522),呈常染色体显性遗传。导致Cole 病的大部分变异都影响编码蛋白中串联生长介素B 样结构域中的半胱氨酸残基[19]。Cole 病临床表现为先天性或早发性点状角化,伴有不规则形状的色素沉着斑,通常见于手臂和腿部,但不见于躯干或肢端区域。
Kotwal等[24]报道了几例单等位基因ENPP1缺陷患者,均未出现低磷血症性佝偻病或全身性婴儿动脉钙化的临床症状,但确实表现出低钙血症或低磷血症,表明ENPP1缺陷可能诱导基因剂量效应影响钙和磷酸盐的稳态。Kato等[9]也发现具有单等位基因ENPP1变异的患者出现ENPP1单倍剂量不足,导致血浆PPi降低,可能是患者出现进行性椎旁韧带骨化症的危险因素。
本研究对发生ENPP1基因变异的2例OPLL患者进行临床表型分析,2 例患者均未发生全身性婴儿动脉钙化,没有身材矮小和佝偻病表型,也未发现点状角化和色素沉着斑等Cole病的表现。患者1血清生化检查发现钙、磷和碱性磷酸酶水平正常,但在C3~C7发生了节段型OPLL,胸椎后纵韧带和黄韧带均有骨化。患者2血清钙水平正常,但血清磷水平在正常低限(0.86 mmol/L),血清碱性磷酸酶水平高于正常值(160 U/L)。患者2发生了C2~C5连续型、C6及C7局灶型、C7~T2 连续型OPLL 及胸椎黄韧带骨化症。与患者1相比较,患者2脊髓压迫程度较重,术前JOA评分仅2.5分。由于患者2携带2个ENPP1变异,患者1仅携带1个ENPP1变异,推测可能是由于变异诱导的剂量效应导致了2例患者临床表型的差异。
通过基因型-临床表型分析,本研究发现ENPP1基因变异造成的OPLL骨化范围广泛、椎管侵占率高、脊髓压迫程度重,造成手术范围广、难度大。ENPP1基因变异的患者可伴有其他方面的异常,如低磷血症、血清碱性磷酸酶水平升高等,因此,在临床上遇到低磷血症、血清碱性磷酸酶水平异常的患者,应考虑进行ENPP1基因分析以明确诊断。在临床前研究中,ENPP1 酶替代疗法已显示出将PPi恢复到正常水平的潜力,从而预防与ENPP1缺乏相关的并发症。据报道,重组ENPP1-Fc 蛋白治疗Enpp1缺陷小鼠可防止异位钙化的发生[25-26]。ENPP1酶替代疗法有望对携带ENPP1基因变异的患者进行治疗,减缓骨化发展。
本研究的局限性:①未对发现ENPP1基因变异的OPLL患者进行PPi水平检测以明确基因变异是否导致PPi水平降低;②在进行WES 的93例患者中,存在各种不同分型的OPLL 病例,由于仅发现2 例ENPP1基因变异患者,本研究并未对ENPP1基因变异患者与其他患者进行临床表型比较。在今后的研究中拟扩大样本量,以阐明不同基因变异对OPLL 临床表型和OPLL分型的影响。
4 结论
ENPP1基因c.T802C(p.Tyr268His)和c.T253C(p.Cys85Arg)变异可能是OPLL患者的致病原因,丰富了ENPP1基因变异谱,为今后OPLL患者应用目前临床开发中的ENPP1酶替代疗法提供了分子诊断依据。
【利益冲突】所有作者均声明不存在利益冲突
