神经胶质细胞钙信号对缺血性脑卒中所致神经功能损伤的影响*
2024-04-07陈钰洁
陈钰洁, 王 磊, 郑 晨
(遵义医科大学珠海校区生理学教研室,广东 珠海 519041)
脑卒中是一种由于脑部血液循环障碍导致局部神经功能缺失的疾病[1]。据《中国心血管健康与疾病报告2021》显示,该疾病仍是导致我国死亡人数最多的疾病,与2009年相比,死亡人数上升了12.4%[2]。其中,有87%为缺血性脑卒中患者[3]。
缺血性脑卒中的病理机制较复杂,目前对于缺血性脑卒中所致的神经功能损伤机制的研究主要涉及细胞缺血性坏死与凋亡、神经组织再灌注损伤、谷氨酸的兴奋性毒性作用、氧化应激损伤、炎症和血脑屏障破坏等方面[4]。近年来研究表明神经胶质细胞在上述损伤机制中均扮演着重要角色[5],而胶质细胞内钙信号可能介导了这些作用,其中,星形胶质细胞是形态和功能最为复杂的胶质细胞[6],参与多种神经系统疾病的发生与发展。因此本文将以星形胶质细胞为重点,对缺血性脑卒中所致的胶质细胞钙信号的激活效应及其机制进行阐述,为实现精准靶向胶质细胞钙信号治疗缺血性脑卒中提供参考资料。
1 胶质细胞种类及基本功能
神经系统主要由神经元与神经胶质细胞这两类细胞所组成,神经元具有电兴奋性,可传导动作电位,而由于神经胶质细胞质膜上电压门控钠离子通道密度极低,且静息状态下K+通透性高,胶质细胞则不具有电兴奋性[7]。胶质细胞主要分为三大类:星形胶质细胞、少突胶质细胞和小胶质细胞。
1.1 星形胶质细胞 星形胶质细胞是一类维持中枢神经系统各级组织稳态并为中枢神经系统提供防御作用的神经胶质细胞[7]。由于星形胶质细胞与神经网络紧密结合,星形胶质细胞还参与其他重要生理功能。在突触及神经发育中,星形胶质细胞通过包围中枢神经系统的突触结构来控制突触的产生、成熟和消除;在形成血脑屏障的过程中,星形胶质细胞参与建立了胶质-神经-血管单位,以此维持血脑屏障结构与功能的稳定;在脑组织代谢方面,星形胶质细胞是大脑中唯一能够储备能量的细胞,并且其终足大量覆盖脑部血管,进而调节脑微循环并为脑组织提供营养与能量等[6,8]。
1.2 少突胶质细胞 少突胶质细胞是一种由少突胶质前体细胞(oligodendrocyte precursor cell, OPC)分化而来、在中枢神经系统中形成髓鞘的神经胶质细胞[9]。在功能上,少突胶质细胞除了显著提升神经传导速度以外,还可以吸收葡萄糖,使其代谢为乳酸和丙酮酸,随后通过单羧酸转运体将乳酸和丙酮酸传递到轴突,为轴突提供能量[10]。在结构上,少突胶质细胞嵌入了一个由大量神经元以及神经胶质细胞所构成的巨大神经网络,因此少突胶质细胞还积极地为神经元提供代谢支持,调节离子和水的动态平衡,并适应活动依赖性神经元信号[11]。
1.3 小胶质细胞 小胶质细胞是中枢神经系统实质中特有的巨噬细胞[8]。小胶质细胞在个体发育的不同阶段表现出不同的功能,在胚胎发育期间,以促进血管发育,去除多余神经元并引导神经元迁移为主;在出生后,主要支持少突胶质前体细胞的生存和发展,确保特定脑区的神经发生,促进棘形成;在髓鞘形成和神经元回路建立完成后,其功能则逐渐转变为保障神经元和少突胶质细胞的存活;当受到干扰时,小胶质细胞将转换为激活状态,增强吞噬作用,产生可溶性因子(如细胞因子、趋化因子等),进而清除细胞碎片并促进细胞再生[8,12]。
2 Ca2+通道的分布及作用机制
细胞内Ca2+作为一种第二信使,通过启动钙调蛋白或丝裂原活化蛋白激酶等介导的信号转导通路,调节细胞生长发育,控制细胞的存活和死亡以及维持细胞功能。神经胶质细胞虽然无法产生并传播动作电位(兴奋),但是它们能通过改变胞浆内Ca2+浓度,从而对神经递质、激素和机械刺激等多种外界刺激做出响应,最终影响神经功能[8,13-14]。神经胶质细胞主要由细胞质膜和细胞器膜上的Ca2+通道、Ca2+转运蛋白共同调节细胞内的钙水平。以下将以星形胶质细胞为重点介绍胶质细胞中主要的Ca2+转运方式(图1)。
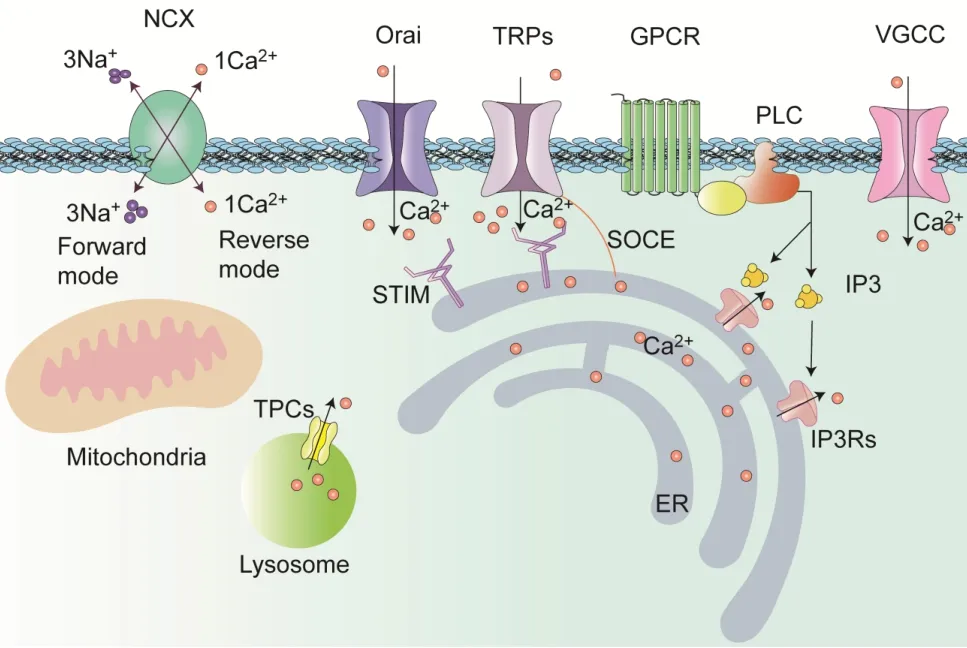
Figure 1. The main source of Ca2+ in neuroglia cells. NCX: so‐dium-calcium exchanger; TRPs: transient receptor potentials; GPCR: G protein-coupled receptor; PLC:phospholipase C; VGCC: voltage-gated calcium chan‐nel; IP3Rs: inositol 1,4,5-trisphosphate receptors;ER: endoplasmic reticulum; SOCE: store-operated Ca2+entry; STIM: stromal interaction molecule; TPCs:two-pore channels.图1 神经胶质细胞内Ca2+的主要来源
2.1 细胞器膜上的Ca2+通道 细胞内钙通道有3 个不同的家族代表:1,4,5-三磷酸肌醇受体(inositol 1,4,5-trisphosphate receptors, IP3Rs)、雷诺丁受体(ryano‐dine receptors, RyRs)和双孔通道(two-pore channels,TPCs)。前两种类型的细胞内通道位于内质网膜上,TPCs 则定位于酸性钙库(溶酶体、高尔基复合体等)[6]。
IP3Rs 和RyRs 是控制Ca2+从钙库释放到胞浆的大型四聚体蛋白,是目前已知最大的离子通道[15]。RyRs 和IP3Rs 在进化上具有相关性,它们均可被胞内Ca2+激活并对内质网腔内的Ca2+浓度同样敏感[15]。当磷脂酶C(phospholipase C, PLC)被G 蛋白偶联受体或受体酪氨酸激酶信号激活时,可形成可溶性IP3分子,该分子与Ca2+共同激活IP3R,从而介导内质网释放Ca2+,在这个过程中,IP3R 的作用被三磷酸腺苷增强,被胞内浓度逐渐增高的Ca2+抑制[16]。已知IP3R 家族包括三个成员:IP3R1、IP3R2 和IP3R3,其中IP3R2 是星形胶质细胞内质网中主要的钙通道,该受体的基因缺失会完全消除或大幅度减少海马区和皮质星形胶质细胞的内质网钙释放和钙信号[17]。
RyRs 也有三种类型:RyR1、RyR2 和RyR3,其中RyR3 仅在脑组织中高表达。但由于该受体主要存在于肌质网上,也被确定为肌肉兴奋-收缩偶联所需的关键钙释放通道,因此,对于该受体的研究主要集中在心律失常、心力衰竭以及骨骼肌的功能障碍等方面[18-19]。
TPCs 被第二信使烟酸腺嘌呤二核苷酸磷酸(nicotinic acid adenine dinucleotide phosphate, NAADP)激活,释放酸性细胞器内的Ca2+[20]。Pereira等[21]报道了星形胶质细胞中也存在TPCs,并且它们的动员与内质网钙库释放的Ca2+有关,事实上,NAADP 引起的星形胶质细胞内的钙信号对内质网钙库耗尽Ca2+也有一定敏感性。
2.2 细胞质膜上的Ca2+通道
2.2.1 钙库操纵性钙通道(store-operated calcium‐channel, SOCC) SOCC 代表了内质网钙含量与质膜钙通透性之间的关联机制,其分子结构包括内质网上的基质相互作用分子(stromal interaction mole‐cule, STIM)和质膜上的Orai 蛋白,两者形成内质网-质膜连接[22]。内质网的Ca2+浓度降低会导致STIM 聚集在内质网-质膜连接处,并在那里进一步结合并激活Orai 蛋白,从而形成钙释放激活的钙内流[23]。当内质网通过内质网钙泵贮存Ca2+时,会导致STIM 和Orai 解离,终止钙内流[23]。这种由Orai1 和STIM1 形成的钙释放激活的钙内流是海马星形胶质细胞Ca2+进入胞内的主要途径,也是引起星形胶质细胞释放胶质递质以调节神经元回路的主要机制[23]。
2.2.2 电压门控Ca2+通道(voltage-gated calcium channels, VGCCs) 神经系统中存在三类主要的VGCCs:Cav1.1-1.4(L 型钙通道)、Cav2.1-2.3(P/Q、N、R 型钙通道)和Cav3.1-3.3(T 型钙通道)[24],并且多种VGCC在星形胶质细胞质膜上均有表达[25]。
2.2.3 瞬时受体电位(transient receptor potential,TRP)通道 TRP 通道是一种能够被化学物质、机械刺激等多种刺激激活的阳离子通道,每一个TRP 通道分子由四个亚基共同组装,形成同质或异质四聚体,根据其氨基酸序列同源性可分为TRPA(an‐kyrin)、TRPC(canonical)、TRPM(melastatin)、TRPML(mucolipin)、TRPP(polycystin)和TRPV(vanilloid)几个亚家族[26-27]。TRP 通道广泛表达于哺乳动物全身细胞和组织,其中TRPC、TRPM 和TRPV 在中枢神经系统中表达水平最高。星形胶质细胞中的TRPC 通道也是SOCC 的组成分子,当内质网储存的Ca2+耗竭时,TRPC 被激活以促进钙内流。星形胶质细胞中的TPRV 被渗透压的变化激活,进而导致大量Ca2+内流以调节细胞容量[23]。
2.2.4 钠钙交换体(sodium-calcium exchanger,NCX) 除了上述Ca2+通道以外,质膜上的NCX 也是Ca2+流入星形胶质细胞内部的另一重要途径。NCX存在两种工作模式,分别为正向工作模式(使Na+流入胞内并将Ca2+挤出)和反向工作模式(促使Ca2+内流)[6]。这两种工作模式的转换主要由星形胶质细胞内Na+浓度以及谷氨酸转运体决定[28]。
3 缺血性脑卒中与神经胶质细胞Ca2+信号失调的关系
3.1 星形胶质细胞 缺血性脑卒中典型的病理生理学特征为脑组织缺血缺氧,可通过对离体脑片进行氧糖剥夺(oxygen-glucose deprivation, OGD)操作来模拟脑卒中后脑组织所处环境。随着OGD 时间的延长,星形胶质细胞内出现钙波,钙波可通过缝隙连接在星形胶质细胞网络中传播并加剧胶质细胞内Ca2+浓度升高[29]。由于星形胶质细胞是合成并大量储存谷氨酸的细胞,所以脑缺血引起星形胶质细胞内Ca2+浓度升高后,会导致星形胶质细胞释放大量谷氨酸,钙依赖性的谷氨酸释放促进突触外N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)的激活,加剧兴奋性毒性导致的脑损伤【23,29-30】。随缺血缺氧加重,脑组织ATP 耗竭会损害星形胶质细胞对谷氨酸的摄取功能,多种途径致使谷氨酸在细胞外大量积累,导致神经元异常兴奋,最终致使神经元功能障碍和死亡[31]。一项后续研究证明在梗死区周围去极化过程中,星形胶质细胞IP3R2基因敲除鼠不仅谷氨酸的释放受到抑制,缺血后的神经元与星形胶质细胞内的钙超载亦有所缓解,梗死体积减小,神经元存活得以维持[32]。另一项研究显示,刺激嘌呤能P2Y 受体(purinergic P2Y receptor,P2YR)也能够激活星形胶质细胞IP3介导的钙释放,然而,该通路可以增强线粒体代谢,进而逆转脑缺血所致的脑组织肿胀以及部分树突状细胞的损伤[33]。该项研究中最值得注意的是,刺激P2YR并未导致星形胶质细胞释放钙依赖性的谷氨酸,与此同时,敲除IP3R2基因还会加剧脑损伤[33]。可见,脑缺血会影响星形胶质细胞内Ca2+浓度,而星形胶质细胞内含有多种Ca2+作用靶点,这些靶点由于其所处的空间位置以及周围Ca2+浓度不同,被激活后出现加剧或减轻脑损伤的作用,这也是IP3介导的钙释放在脑卒中的疾病进程中拥有双重作用的主要原因。
在缺血性脑卒中急性期,脑组织中凝血酶原的合成增加,产生的凝血酶激活星形胶质细胞膜上的蛋白酶激活受体(protease-activated receptor, PAR),PAR 通过G 蛋白偶联受体激活PLC,进而诱发IP3 介导的内质网腔的Ca2+释放;与此同时,凝血酶还会导致内质网局部STIM1 的重组,触发钙库操纵性钙内流(store-operated Ca2+entry, SOCE),上述两种方式使星形胶质细胞内Ca2+浓度成倍增加,最终加剧脑损伤[34-35]。不仅脑组织自身产生的凝血酶会对脑卒中的存活产生不利影响,缺血性脑卒中还会造成血脑屏障破坏,导致血浆中的凝血酶原渗漏到脑实质,并裂解成凝血酶,凝血酶诱导星形胶质细胞膜上的TRPC3 表达,启动SOCE 机制,胞内大量的Ca2+导致严重的星形胶质细胞增生症,进而造成脑卒中神经功能损伤的不良预后[36]。不仅如此,星形胶质细胞上TRPV 家族亦参与了脑卒中的进展,TRPV1 通过非受体络氨酸炎症信号通路诱导星形胶质细胞发生反应,刺激白细胞介素1β 等炎症子的分泌,导致脑卒中后炎症持续发展[37]。
此外,缺血性脑损伤后,星形胶质细胞启动血红素加氧酶1/一氧化碳(carbon oxide, CO)通路,CO 通过增加L 型钙通道介导的Ca2+内流,并同时增强低氧诱导因子依赖性血管内皮生长因子(vascular endo‐thelial growth factor, VEGF)的启动子活性,进一步诱导星形胶质细胞内VEGF 的表达,以此维持和修复神经血管耦合功能,为损伤区域提供氧气和营养[38]。同时,在缺血过程中激活神经元上的TRPC6 可防止神经元死亡,而阻断TRPC6 则会增加其对缺血的敏感性[39],不利于神经元的存活。综上所述,星形胶质细胞中的各种Ca2+通道介导的Ca2+内流在脑卒中进展的作用不仅是加剧脑损伤,还在各方面对神经元提供保护作用,这也提示研究在治疗脑卒中的过程中准确使用特异性Ca2+通道阻滞剂是有价值的。然而,脑卒中这些Ca2+通道被激活的具体时间点以及不同Ca2+通道相互之间的联系,还需要进一步探索,以此实现对脑卒中后Ca2+通道进行更精确地调控以减少其对脑组织的损伤。
3.2 少突胶质细胞 少突胶质细胞内Ca2+浓度升高方式和星形胶质细胞类似,但对机体的调控作用区别很大。蒂莫西综合征(Timothy syndrome, TS)是一种由于编码Cav1.2 通道的基因突变致使细胞Ca2+过度内流而导致多器官结构和功能障碍的疾病。Cheli等[40]利用TS 小鼠研究L 型Ca2+通道在脑中的作用,从TS 小鼠皮质分离的OPC 检测到更多的L 型Ca2+内流,同时证明了这种Ca2+内流可促进OPC 的发育,加快少突胶质细胞的形成与成熟,并且借助钙/钙调蛋白依赖的蛋白激酶Ⅱ促进髓鞘的形成。
研究表明小脑白质缺血引起的钠泵抑制和谷氨酸释放会诱发代谢变化,从而造成少突胶质细胞酸化,激活氢离子门控的TRP 通道,Ca2+通过该通道进入少突胶质细胞,最终导致髓鞘损伤[41]。在脑组织急性缺血时,中枢神经系统白质轴突大量释放谷氨酸,从而过度激活NMDAR 介导的髓鞘细胞毒性作用[42]。除此以外,研究证实Ca2+升高对于髓鞘也有正向作用,其作用涉及驱动髓鞘的延长,参与调节细胞骨架的生长和髓鞘蛋白的组装,然而,低于一定Ca2+瞬变速率或者高钙负荷的长时程Ca2+爆发则会导致髓鞘缩短[43]。
3.3 小胶质细胞 小胶质细胞作为大脑的免疫活性细胞拥有多种表型,并能够表达多种受体,这样有助于小胶质细胞对不同的机体环境变化信息做出特异性的反应。在生理过程中,小胶质细胞对中枢神经系统进行免疫监视,调控突触的形成与修剪;而病理情况下,小胶质细胞能够通过释放多种细胞因子介导炎症反应或者进行神经修复[44-45],而小胶质细胞中的Ca2+信号在很大程度上控制着这些不同的功能。
脑卒中所致的脑皮层扩散性抑制能够诱导小胶质细胞Ca2+内流,并且随着卒中时间增加,小胶质细胞内的Ca2+活动更为活跃[46]。在急性期,细胞表面嘌呤能受体和SOCE 机制共同介导小胶质细胞Ca2+内流,进而刺激NMDAR 介导的Ca2+进入神经元,使离子稳态失调进一步放大,增加梗死面积[46-47]。此外,小胶质细胞上的嘌呤能受体还可以启动P2YR-IP3通路,促使小胶质细胞和神经元之间形成嘌呤能连接,这些连接能够有效避免过量的Ca2+流入病灶区神经元内[48]。然而,随着缺血时间延长,IP3 介导的钙库耗尽,会激活SOCC,使细胞外Ca2+大量内流,这种延迟的Ca2+内流影响了一系列信号通路,例如钙调磷酸酶通路,该通路参与调节免疫细胞中的基因表达,导致炎性细胞因子的持续产生,使小胶质细胞的作用从神经保护转变为神经毒性[47]。
4 结论与展望
胶质细胞中钙信号增加不仅有加剧脑损伤的作用,亦有保护脑组织的作用,这可能与胶质细胞上钙受体亚型、钙信号作用靶点以及脑卒中的时间进程等因素有关。神经元、少突胶质细胞和OPC 都易受缺血性休克的影响,而星形胶质细胞在卒中后有更强的适应和恢复能力,一方面它可以有效对抗谷氨酸兴奋性毒性;另一方面星形胶质细胞可以清除活性氧,此外,在氧气和葡萄糖缺乏的情况下,星形胶质细胞也能为神经元提供能量,并增强星形胶质细胞及其周围神经元的抵抗力[49]。然而,当脑组织严重损伤或缺血时间过长时,星形胶质细胞由于胞内Ca2+浓度异常增高,进而转变为神经元胞外谷氨酸的主要来源;梗死区星形胶质细胞中也会产生异常Ca2+波,进一步引发缺血病灶以外的区域释放谷氨酸,从而促进梗死面积的扩展[30-31]。这些研究证据提示了胶质细胞钙信号相关通路有望成为脑卒中治疗的潜在靶点。
目前,各国学者进行了大量Ca2+通道阻滞剂对缺血性卒中作用的临床研究,观察到L 型Ca2+通道阻滞剂能有效缓解神经功能障碍,阻止早期较小的脑梗塞区域的扩大[50]。然而,也有临床试验结果表明Ca2+通道阻滞剂对于脑卒中患者的疾病结局没有明显影响[51]。因此,还需要更多临床前实验对缺血性脑卒中Ca2+信号机制进行更加详细的研究,以详尽阐明胶质细胞Ca2+信号在缺血性脑卒中疾病进程中的作用以及能够靶向性控制信号通路中的受体,从而实现对于Ca2+信号更精准的调控,以此为缺血性脑卒中治疗提供新选择。
